
Tin tức
Tế bào gốc ung thư P1
Tác giả Oleg Shuvalov,*ORCID,Yulia Kirdeeva,Alexandra Daks,Olga Fedorova,Sergey Parfenyev,Hans-Uwe Simon, và Nickolai A. Barlev*
Biên dịch: Hoàng Đôn Hòa | Viện Y học bản địa Việt Nam
Tái lập trình chuyển hóa ung thư là một quá trình phức tạp giúp tế bào ác tính có lợi thế chọn lọc để phát triển và nhân lên trong môi trường khắc nghiệt do hệ miễn dịch tạo ra. Quá trình này củng cố sự tăng sinh, xâm lấn, phòng thủ chống oxy hóa của ung thư và kháng lại khả năng miễn dịch và điều trị ung thư. Không có gì ngạc nhiên khi việc tái cấu trúc chuyển hóa được coi là một trong những “Đặc điểm của ung thư”. Đáng chú ý, quá trình này thường bao gồm nhiều con đường bổ sung và chồng chéo. Ngày nay, người ta đã biết rằng việc ức chế có chọn lọc cao chỉ một trong các con đường trong tế bào khối u thường dẫn đến đáp ứng hạn chế và sau đó là sự xuất hiện của kháng thuốc. Do đó, để tăng hiệu quả tổng thể của thuốc chống khối u, nên sử dụng các tác nhân đa mục tiêu có thể đồng thời ức chế một số quá trình quan trọng trong tế bào khối u. Bài đánh giá này tập trung vào một nhóm các hợp chất tự nhiên có nguồn gốc thực vật đồng thời nhắm mục tiêu các con đường khác nhau của quá trình chuyển hóa liên quan đến ung thư, bao gồm đường phân hiếu khí, hô hấp, glutaminolysis, chuyển hóa một cacbon, tạo lipogenesis de novo và quá trình oxy hóa β của axit béo. Chúng tôi chỉ thảo luận về những hợp chất thể hiện hoạt tính ức chế đối với một số con đường trao đổi chất cũng như một số con đường tín hiệu quan trọng trong ung thư. Thông tin về dược động học của chúng ở động vật và người cũng được trình bày. Tóm lại, một số hợp chất có nguồn gốc thực vật đã biết có thể nhắm mục tiêu nhiều con đường chuyển hóa và tín hiệu trong các bệnh ác tính khác nhau, điều này mang lại tiềm năng lớn để cải thiện hơn nữa liệu pháp chống ung thư.
1. Giới thiệu
Theo thống kê của Tổ chức Y tế Thế giới, ung thư là nguyên nhân gây tử vong đứng thứ hai trên toàn cầu. Theo thống kê ung thư toàn cầu (GLOBOCAN), năm 2020, có 19,3 triệu ca ung thư mới và gần 10 triệu ca tử vong liên quan đến ung thư trên toàn thế giới. Do đó, ngoài các phương pháp điều trị ung thư truyền thống đã được thiết lập, vẫn luôn cần tìm kiếm và phát triển các liệu pháp chống ung thư mới để giải quyết thách thức này.
Để điều trị ung thư, chúng ta cần nhắm mục tiêu vào những đặc điểm riêng biệt phân biệt tế bào ung thư với tế bào bình thường. Điều này là cần thiết để giảm thiểu tác dụng phụ và bảo vệ các mô và cơ quan khỏe mạnh khỏi tác động của các thuốc hóa trị gây hại.
Một trong những “Đặc điểm của ung thư” được công nhận rộng rãi hiện nay trong bối cảnh điều trị là sự tái lập trình và tính dẻo của quá trình chuyển hóa. Tế bào ung thư có nguồn gốc khác nhau được đặc trưng bởi một tập hợp các thay đổi chuyển hóa và tính dẻo, cung cấp năng lượng và khả năng thích ứng cho tế bào ác tính. Những đặc điểm chuyển hóa cụ thể này là mục tiêu thích hợp để can thiệp điều trị.
Ngày nay, người ta chấp nhận rộng rãi rằng việc ức chế chỉ một trong các quá trình trong tế bào khối u, ngay cả bằng một loại thuốc có độ đặc hiệu cao, thường dẫn đến đáp ứng hạn chế và sau đó là sự xuất hiện của kháng thuốc. Để tăng hiệu quả tổng thể của liệu pháp chống ung thư, nên sử dụng một số loại thuốc, hoặc thuốc đa mục tiêu, có thể đồng thời ức chế một số quá trình quan trọng trong tế bào khối u. Tuy nhiên, phần lớn các thuốc tổng hợp chống ung thư hiện đại đều có tác dụng phụ và khả năng dung nạp/kháng thuốc đa năng.
Trong thập kỷ qua, sự quan tâm đến việc sử dụng tiềm năng y học của các hợp chất tự nhiên chống lại ung thư đã tăng lên đáng kể, và số lượng các ấn phẩm liên tục tăng (Hình 1).
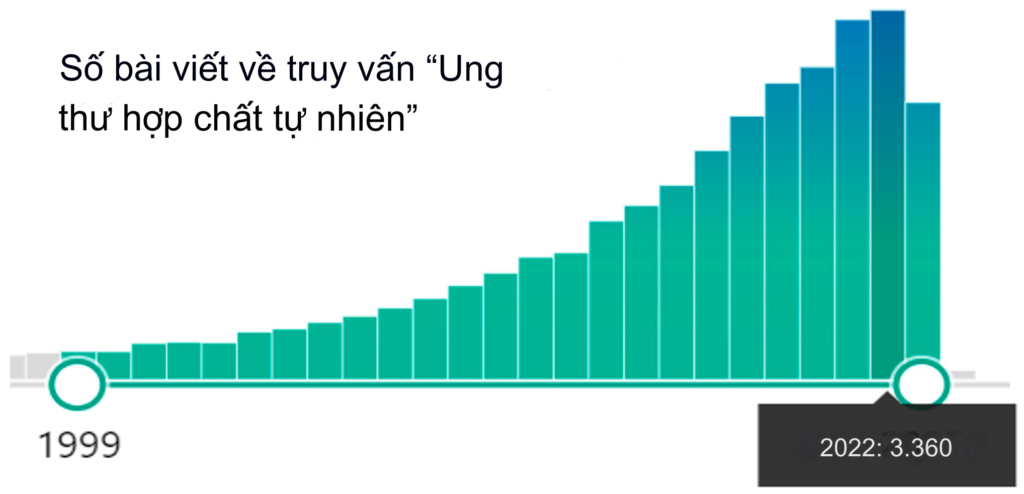
Thật vậy, các hợp chất tự nhiên, cùng với các hóa chất tổng hợp, có thể hữu ích trong điều trị ung thư. Cách tiếp cận này hoàn toàn hợp lý bởi vì hầu hết các loại thuốc hóa trị được sử dụng thường xuyên đều có nguồn gốc từ thực vật và actinomycetes: paclitaxel, vincristine, vinblastine, doxorubicin, camptothecin, etoposide, topo- và irinotecan, v.v. Ví dụ, hành tinh của chúng ta chứa khoảng 391.000 loài thực vật, tạo ra hàng chục nghìn hợp chất hóa học với nhiều hoạt tính sinh học khác nhau, bao gồm cả hoạt tính chống ung thư. Hơn nữa, nó cung cấp rất nhiều hợp chất ứng cử viên với nhiều nguồn và cấu trúc mới lạ.
Thứ hai, một số hợp chất tự nhiên từ nhóm thực phẩm chức năng có thể tiêu diệt tế bào ung thư và hỗ trợ điều trị ung thư như một phần của phác đồ hóa trị hiện đại.
Thứ ba, một số thực phẩm chức năng như curcumin, resveratrol, quercetin, ginsenosides, 20-hydroxyecdysone và các loại khác không chỉ có nhiều hoạt tính chống ung thư mà còn thể hiện các đặc tính dược lý (chống oxy hóa, chống tiểu đường, chống viêm, bảo vệ gan và thần kinh, v.v.) rất có lợi cho bệnh nhân ung thư đang hóa trị.
Y học cổ truyền Trung Quốc, Ayurveda, Kampo và các hệ thống y học cổ truyền khác sử dụng các loại thảo mộc và công thức được xác định theo kinh nghiệm qua nhiều thế kỷ, đã được chứng minh là có hiệu quả trong các nghiên cứu tiền lâm sàng và lâm sàng. Các nghiên cứu chuyên sâu về các chất được sử dụng rộng rãi trong y học dân tộc đã dẫn đến việc phân lập một số hợp chất có đặc tính sinh học hữu ích, bao gồm cả đặc tính chống khối u. Một số trong số chúng ta thường xuyên tiêu thụ dưới dạng thực phẩm, đồ uống, gia vị hoặc thực phẩm bổ sung.
Một số hợp chất tự nhiên nhắm vào mục tiêu tái lập trình chuyển hóa đã được biết đến và được tóm tắt trong một số bài đánh giá xuất sắc.
Tuy nhiên, ở đây chúng tôi xem xét một tập hợp các hợp chất tự nhiên đồng thời đáp ứng ba tiêu chí: (1) ức chế một số con đường chuyển hóa liên quan đến ung thư; (2) có thông tin về dược động học và sinh khả dụng của chúng; (3) hợp chất thường thể hiện hồ sơ an toàn hoặc/và đã được người dân tiêu thụ hoặc sử dụng trong nhiều thử nghiệm lâm sàng.
Các tế bào ác tính có khả năng thích ứng tuyệt vời và có thể thích ứng với sự ức chế các con đường sinh hóa hoặc tín hiệu nhất định thông qua các con đường bỏ qua và phản vệ được điều chỉnh tốt. Do đó, chúng tôi chỉ tập trung vào một nhóm các hợp chất tự nhiên có nguồn gốc thực vật đồng thời nhắm mục tiêu các khía cạnh khác nhau của quá trình tái lập trình chuyển hóa, bao gồm đường phân hiếu khí, hô hấp, glutaminolysis, chuyển hóa một cacbon, tạo lipogenesis de novo và quá trình oxy hóa β của axit béo.
Để thực hiện khảo sát, chúng tôi đã thu thập thông tin từ các cơ sở dữ liệu có sẵn (MEDLINE/PubMed, Google Scholar, Web of Science, Scopus, Elsevier, SpringerLink và Wiley Online Library).
Ngoài tác động tiêu cực của chúng đối với một số con đường sinh hóa, tất cả các hợp chất được xem xét cũng nhắm mục tiêu các con đường tín hiệu khác nhau, bao gồm PI3K/AKT/mTOR, ERK/MAPK, Jac/STAT, v.v. Các đặc tính này giúp tăng cường khả năng đa mục tiêu của chúng, có thể làm tăng hiệu quả của liệu pháp chống ung thư. Hơn nữa, nhiều loại trong số chúng còn có các đặc tính dược lý có lợi khác, bao gồm chống oxy hóa, hạ đường huyết, bảo vệ gan và thần kinh, có thể cực kỳ hữu ích khi can thiệp hóa trị để giảm hậu quả có hại của nó đối với các mô không ung thư. Điều rất quan trọng đối với y học chuyển đổi, độ an toàn và sinh khả dụng của các hợp chất được đánh giá cũng được thảo luận.
2. Tái lập trình Chuyển hóa trong Ung thư
Như đã đề cập ở trên, tái lập trình chuyển hóa được coi là một trong những “Đặc điểm của ung thư”. Để giải quyết vấn đề này như một mục tiêu đa mục tiêu cho các hợp chất tự nhiên, dưới đây, chúng tôi thảo luận ngắn gọn về các thuộc tính chính của việc chuyển đổi chuyển hóa và vai trò của các gen ung thư và các con đường tín hiệu trong hiện tượng phức tạp này.
2.1. Tăng Đường phân (“Hiệu ứng Warburg”)
Đường phân bị rối loạn là một “Đặc điểm của ung thư” quan trọng và còn được gọi là hiệu ứng “Warburg”. Hiệu ứng này ngụ ý rằng các tế bào ung thư duy trì mức độ đường phân cao ngay cả trong điều kiện normoxic. Điều này có nghĩa là các khối u khác nhau sử dụng glucose nhiều hơn các tế bào bình thường do tăng biểu hiện các chất vận chuyển glucose (ví dụ: GLUT1) và một số enzym đường phân.
Tăng cường đường phân là rất phổ biến trong các khối u đến nỗi nó đã tạo thành cơ sở cho phương pháp phát hiện cả khối u nguyên phát và thứ phát trong cơ thể bằng PET/CT. 18F-Fluorodeoxyglucose (FDG) là một chất tương tự glucose được vận chuyển qua các chất vận chuyển glucose vào tế bào ung thư sau đó được phosphoryl hóa qua trung gian hexokinase 2 (HK2). Do đó, các khu vực tăng trưởng ác tính được phát hiện dựa trên sự gia tăng hấp thu và sử dụng glucose.
Như đã đề cập ở trên, glucose được vận chuyển vào tế bào ác tính thông qua một số chất vận chuyển glucose (GLUT); GLUT1 và GLUT3 được coi là những chất chính. Các chất vận chuyển này, đặc biệt là GLUT1, thường được điều hòa tăng cường trong các khối u khác nhau và thúc đẩy khả năng xâm lấn và kháng trị liệu của chúng. Việc ức chế các chất vận chuyển này, có thể làm giảm quá trình đường phân không kiểm soát được, là một phương pháp chống ung thư đang phát triển với một số hợp chất đang được nghiên cứu trong các mô hình tiền lâm sàng và lâm sàng.
Khi vào tế bào, glucose trải qua một loạt các phản ứng enzyme để tạo thành hai phân tử pyruvate làm sản phẩm cuối cùng (Hình 2). Trong phản ứng đầu tiên, glucose được hoạt hóa bằng cách phosphoryl hóa có nguồn gốc từ hexokinase (HK). Đây là bước giới hạn tốc độ đầu tiên của quá trình đường phân. Hơn nữa, bên cạnh đường phân, sản phẩm của phản ứng này, glucose-6-phosphate, được chuyển hóa trong các con đường sinh tổng hợp glycogen, pentose phosphate và hexosamine, có nghĩa là nó đóng vai trò quan trọng trong tổng hợp ATP, dự trữ glucose, làm giàu NADH và glycosyl hóa protein, tương ứng. Trong số bốn đồng dạng HK được xác định cho đến nay, vai trò gây ung thư của HK2 được công nhận rộng rãi. Bên cạnh vai trò chính trong quá trình đường phân, HK2 có thể liên kết với kênh anion phụ thuộc điện áp (VDAC) trên màng ngoài ty thể, nơi nó ức chế hoạt động của các protein gây chết theo chương trình của họ Bcl-2 và bảo vệ tế bào khối u khỏi các kích thích tử vong. Nói chung, HK2 đóng một vai trò quan trọng trong quá trình gây ung thư theo nhiều cách và là mục tiêu thuốc mong muốn cho liệu pháp điều trị ung thư.
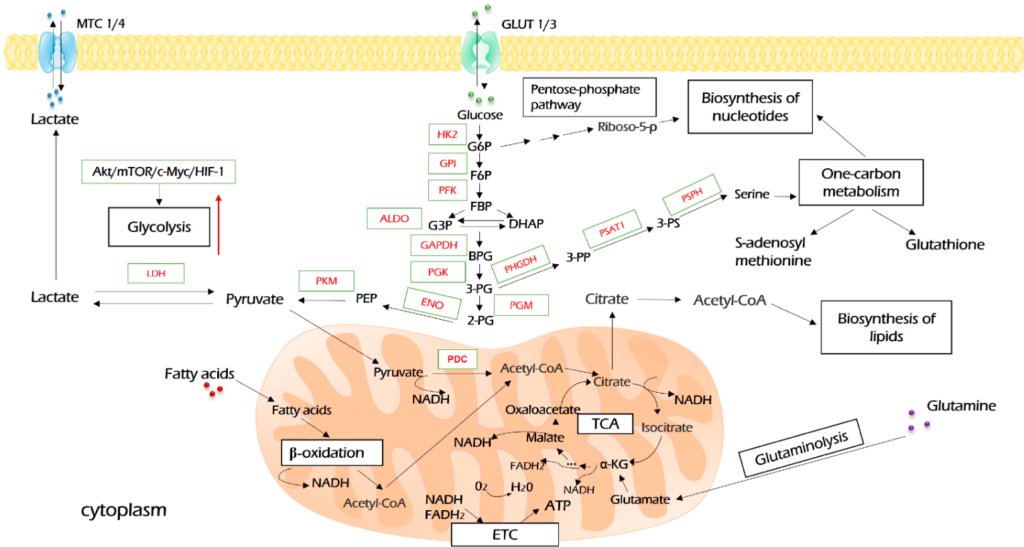
Bước giới hạn tốc độ thứ hai của quá trình đường phân được thực hiện bởi phosphofructokinase (PFK), xúc tác chuyển đổi fructose 6-phosphate thành fructose 1,6-bisphosphate. PFK1 có ba đồng dạng đặc trưng cho mô: tiểu cầu (PFKP), cơ (PFKM) và gan (PFKL), tất cả đều có thể được biểu hiện quá mức trong các khối u khác nhau. Ngoài các chức năng trao đổi chất rất quan trọng trong tế bào khối u, PFK còn tham gia vào một số con đường tín hiệu, ví dụ như hỗ trợ tín hiệu PI3K, YAP/TAZ và β-catenin.
Bước đường phân giới hạn tốc độ thứ ba được xúc tác bởi pyruvate kinase (PK). Đồng dạng nổi bật nhất liên quan đến ung thư là PKM2. Enzyme này xúc tác bước đường phân cuối cùng và trải qua một quy định allosteric phức tạp. PKM2 điều phối dòng carbon giữa đường phân, phosphoryl hóa oxy hóa, chuyển hóa một cacbon và glutaminolysis. Ngoài quá trình trao đổi chất, PKM2 thúc đẩy quá trình tạo khối u và kháng hóa chất bằng nhiều cơ chế, bao gồm kích hoạt HIF1α, c-Myc, STAT3 và Oct-4. Có lẽ không ngạc nhiên khi việc ức chế dược lý PKM2 là rất mong muốn và là chủ đề của nhiều thử nghiệm lâm sàng.
Hơn nữa, ở các tế bào bình thường, pyruvate được nhập khẩu vào ty thể và đi vào chu trình TCA để hỗ trợ quá trình phosphoryl hóa oxy hóa (OXPHOS). Ngược lại, trong các tế bào ung thư, một lượng quá mức pyruvate được tổng hợp do sự điều hòa tăng đáng kể của quá trình đường phân, có thể làm chậm quá trình này. Trong trường hợp này, pyruvate dư thừa được chuyển đổi thành lactate bởi lactate dehydrogenase (LDH). Hấp thu glucose cao và sản xuất lactate là hai dấu hiệu nổi bật của quá trình chuyển hóa ung thư.
Tất cả các đồng dạng LDH, nhưng đặc biệt là LDHA, thúc đẩy các đặc tính ác tính khác nhau và thúc đẩy các quá trình gây ung thư chính. Chúng làm tăng những thay đổi chuyển hóa liên quan đến ung thư, tăng cường sự phát triển, tiềm năng di căn và khả năng kháng trị liệu, làm giảm khả năng miễn dịch chống khối u, v.v. Mức độ hoạt động của LDH trong huyết thanh cao được các bác sĩ ung thư biết đến như một dấu hiệu mạnh mẽ của tiên lượng xấu và đáp ứng với liệu pháp điều trị.
Bản thân lactate là một hợp chất độc hại và dẫn đến quá trình axit hóa tế bào chất. Do đó, để khắc phục độc tính của nó, lactate nên được xuất ra bên ngoài tế bào. Quá trình này được thực hiện bởi MCT và dẫn đến quá trình axit hóa không gian nội bào xung quanh các tế bào khối u. Trong số bốn đồng dạng MCT, MCT1 và MCT4 là những đồng dạng được biểu hiện chủ yếu trong ung thư và đã được xác định là mục tiêu điều trị tiềm năng. Điều thú vị là MCT không chỉ tham gia vào quá trình bài tiết lactate mà còn có thể nhập khẩu lactate trong các tế bào ác tính phụ thuộc OXPHOS, nguyên bào sợi liên kết với ung thư (CAF) hoặc các tế bào khác của vi môi trường khối u để oxy hóa tiếp theo, điều này thúc đẩy quá trình cộng sinh trao đổi chất bên trong khối u. MCT thường được biểu hiện quá mức trong các khối u ác tính; chúng tạo điều kiện cho sự hình thành di căn và tạo mạch. Liệu pháp nhắm mục tiêu MCT đang trải qua các thử nghiệm tiền lâm sàng và lâm sàng.
Thoạt nhìn, việc tăng cường đường phân trong tế bào ung thư dường như là một nghịch lý vì đường phân không phải là một quá trình hiệu quả để sản xuất ATP. Về lý thuyết, chỉ có hai phân tử ATP được tạo ra trên mỗi phân tử glucose khi đường phân, thay vì 38 phân tử ATP được tạo ra bởi OXPHOS như một sự tiếp nối của đường phân. Tuy nhiên, có một số lý do để chọn con đường này hơn OXPHOS, một số trong số đó được liệt kê dưới đây.
Đường phân cho phép các tế bào ung thư tổng hợp ATP nhanh chóng. Hơn nữa, nó thúc đẩy dòng chảy vào các con đường sinh tổng hợp. Sản phẩm trung gian của quá trình đường phân, 3-phosphoglycerate, có thể được chuyển đổi thành serine trong ba bước bởi PHGDH, PSPH và PSAT1, mở ra cánh cổng cho quá trình chuyển hóa một cacbon và sinh tổng hợp nucleotide. Điều này có thể được xem như một cầu nối đồng hóa liên kết sự đồng hóa glucose với quá trình chuyển hóa một cacbon. Citrate, có nguồn gốc từ pyruvate trong chu trình TCA, là nguồn tạo lipogenesis và sinh tổng hợp một số axit amin. Hơn nữa, quá trình axit hóa qua trung gian đường phân làm bất hoạt phản ứng miễn dịch chống ung thư và tinh chỉnh vi môi trường khối u. Cuối cùng, đường phân có thể tác động đến các tín hiệu truyền tín hiệu thông qua các chất trung gian của nó có các đặc tính của phân tử tín hiệu (ví dụ: fructose 1,6-bisphosphate và lactate).
Do đó, đường phân được điều chỉnh bởi một số gen ung thư. Trong số những người khác, các chất điều hòa chính của nó là c-Myc, HIF1α, AKT và mTOR, có thể hoạt hóa các gen đường phân (c-Myc, HIF1α) hoặc có thể điều chỉnh trực tiếp và gián tiếp hoạt động của các enzym thông qua các sửa đổi sau dịch mã và tương tác protein-protein.
Ngoài việc tăng mức độ tăng sinh và di căn, glucose cao sẽ khởi đầu sự mất ổn định bộ gen và các đột biến de novo, bao gồm KRASG12D, trong các tế bào tuyến tụy không gây ung thư. Ngoài ra, lượng đường glucose cao có thể dẫn đến mất cân bằng nucleotide và ức chế quá trình sửa chữa cắt bỏ nucleotide (NER).
2.2 Chu trình TCA và OXPHOS
Pyruvate liên kết đường phân với hô hấp. Nó được nhập khẩu vào ty thể và oxy hóa bởi PDK thành acetyl-CoA (Hình 3). Acetyl-CoA là nguồn chính cho cả quá trình tạo lipogenesis (quá trình hình thành chất béo) và chu trình Krebs (TCA) để cung cấp nhiên liệu cho OXPHOS. TCA là một trung tâm phân phối lại các nguồn carbon để tạo ra năng lượng tế bào và là tiền chất cho các con đường sinh tổng hợp liên kết đường phân, glutaminolysis, sinh tổng hợp và quá trình oxy hóa beta của axit béo, hô hấp và chuyển hóa axit amin vào mạng lưới chuyển hóa (Hình 2). Các chất trung gian của nó là citrate và α-ketoglutarate (α-KG). α-KG có thể được bắt nguồn từ glutamine khi glutaminolysis và sau đó có thể được carboxyl hóa khử để tạo thành citrate, cung cấp năng lượng cho chu trình TCA và OXPHOS. Quá trình này được gọi là quá trình đồng hóa.
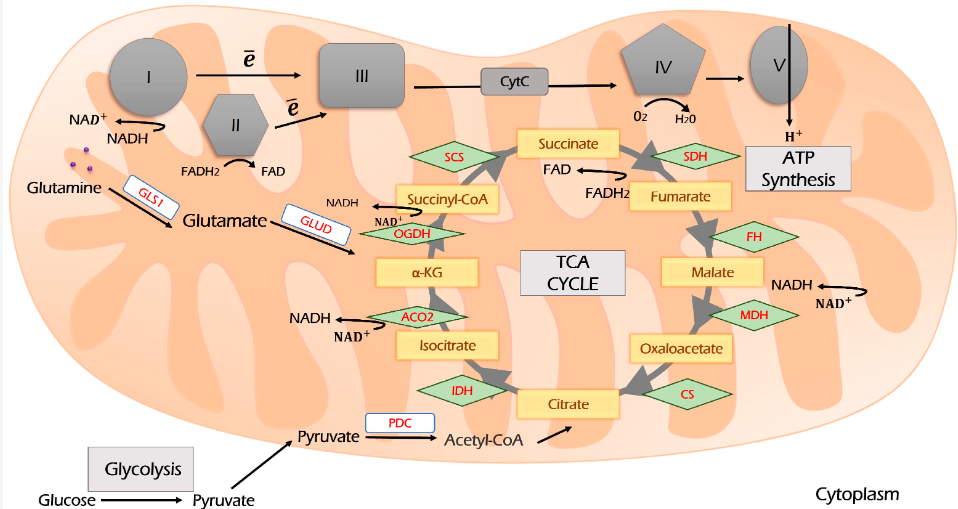
Quá trình đồng hóa glutamine xảy ra trong ty thể và được gọi là “glutaminolysis”. Glutamine được vận chuyển đến ty thể bởi biến thể SLC1A5. Glutaminolysis được xúc tác bởi glutaminase, được mã hóa bởi hai đồng dạng: GLS1 và GLS2. Glutaminolysis liên quan đến ung thư được liên kết với GLS1, được biểu hiện quá mức bởi nhiều loại ung thư.
Sau đó, glutamate có thể được chuyển đổi thành α-KG bởi GLUD (GDH), lần lượt, cung cấp năng lượng cho chu trình TCA và OXPHOS hoặc trở thành cơ chất cho transaminase, bao gồm glutamic pyruvic transaminase 2 (GPT2), glutamic oxaloacetic transaminase 2 (GOT2) , hoặc PSAT1 để sản xuất các axit amin không thiết yếu (lần lượt là alanine, aspartate và phosphoserine).
Hơn nữa, glutamate thúc đẩy quá trình sinh tổng hợp glutathione tripeptide, đóng vai trò quan trọng trong quá trình cân bằng nội môi oxi hóa khử và làm trung gian bảo vệ chống oxy hóa. Trong tế bào chất, glutamine có thể được chuyển đổi thành aspartate bằng asparagine synthetase (ASNS). Cuối cùng, cả glutamine và aspartate có nguồn gốc từ glutamine đều là chất cho cacbon và nitơ cho quá trình sinh tổng hợp nucleotide pyrimidine và purine.
Cũng như bất kỳ quá trình trao đổi chất nào khác bị ung thư làm thay đổi, quá trình chuyển hóa glutamine được kiểm soát bởi một số gen ung thư và gen ức chế khối u. Ví dụ, c-Myc thúc đẩy quá trình hấp thu glutamine bằng cách hoạt hóa các gen mã hóa chất vận chuyển glutamine SLC1A5. Hơn nữa, nó tăng cường biểu hiện của GLS1 thông qua việc ức chế các chất điều hòa âm tính của nó: miR-23a, miR-23b và lncRNA GLS-AS. Nó tăng cường biểu hiện của GLUD1, GPT2, GOT1, GOT2 và PSAT1. mTORC1 điều hòa tăng GLS1 bằng cách tăng biểu hiện c-Myc. Trong ung thư biểu mô tuyến tụy, KRAS lập trình lại quá trình chuyển hóa glutamine thành quá trình tổng hợp aspartate có nguồn gốc từ glutamine, sản xuất NADPH và cân bằng cân bằng nội môi oxi hóa khử tế bào với quá trình tổng hợp đại phân tử.
Do đó, glutamine là một chất cho cacbon và nitơ nổi bật, cung cấp cả năng lượng sản xuất thông qua việc xâm nhập vào TCA và OXPHOS và cung cấp năng lượng cho các quá trình sinh tổng hợp để sản xuất axit béo, axit amin không thiết yếu, glutathione và cả nucleotide pyrimidine và purine. Việc ức chế GLS1 và GDH và ức chế quá trình chuyển hóa glutamine được công nhận là một phương pháp chống ung thư quan trọng. Nó ức chế sự phát triển của ung thư, di căn và hô hấp ty thể và ức chế tế bào gốc ung thư.
2.4 Chuyển hóa Lipid
Ngoài hiện tượng Warburg và tăng glutaminolysis, chuyển hóa lipid cũng trải qua quá trình tái lập trình chuyển hóa toàn diện trong tế bào ung thư. Nói chung, nó bao gồm quá trình hấp thụ axit béo (FA), sinh tổng hợp lipid de novo (lipogenesis) và quá trình oxy hóa β của axit béo (FAO). Tất cả các quá trình này có liên quan đến sự hình thành khối u và thúc đẩy sự tăng sinh, di chuyển, xâm lấn và kháng thuốc của các tế bào ác tính cũng như điều chỉnh sự tương tác của chúng với vi môi trường.
Axit béo có thể xâm nhập vào tế bào ung thư bằng cách khuếch tán hoặc bằng cách được nhập khẩu bởi các protein vận chuyển FA (Hình 5). Các protein vận chuyển FA được biểu thị bằng FATP1-6 (protein vận chuyển axit béo 1-6), FABP1-9 (protein liên kết axit béo 1-9) và chất vận chuyển axit béo CD36. Bên trong tế bào, các axit béo liên kết thuận nghịch với FABP, hoạt động như các chất hỗ trợ lipid nội bào.
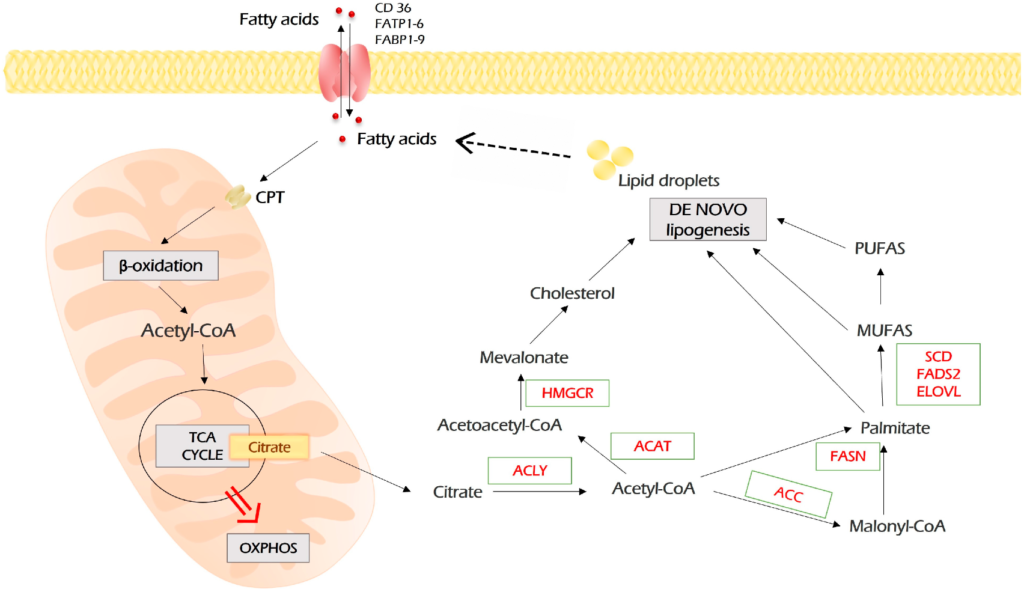
Khi vào tế bào, axit béo trải qua quá trình liên hợp hoạt hóa với coenzyme A, được thực hiện bởi acyl-CoA synthetases (ACSS, ACSM, ACSL). Sau đó, đối với quá trình oxy hóa β, axit béo acyl-CoA cần phải đi vào ty thể. Bước giới hạn tốc độ này được xúc tác bởi carnitine palmitoyltransferases (CPT1 và CPT2) lần lượt được nội địa hóa trên màng ngoài và màng trong ty thể.
Trong ty thể, trong quá trình oxy hóa β, axit béo acyl-CoA được phân cắt thành acetyl-CoA bằng một chu trình bốn bước lặp lại được xúc tác bởi bốn enzyme. Sản phẩm cuối cùng, acetyl-CoA, đi vào chu trình TCA, tiếp theo là quá trình phosphoryl hóa oxy hóa để tạo ra ATP.
Quá trình tạo lipogenesis de novo bắt đầu với acetyl-CoA, là “khối xây dựng” cho tất cả các axit béo. Nguồn chính của acetyl-CoA là quá trình khử carboxyl oxy hóa pyruvate, xảy ra sau quá trình đường phân. Ngoài ra, acetyl-CoA có thể được sản xuất từ citrate khi glutaminolysis và quá trình oxy hóa β của axit béo. Citrate được chuyển đổi thành acetyl-CoA bởi ATP citrate lyase (ACLY). Sau đó, acetyl-CoA carboxylase (ACC) xúc tác quá trình chuyển đổi acetyl-CoA và một phân tử bicarbonate thành malonyl-CoA. Hơn nữa, axit béo synthase (FASN) đã xúc tác quá trình tổng hợp palmitate từ malonyl-CoA và acetyl-CoA (Hình 5).
Quá trình hấp thu, lưu trữ và sử dụng lipid là một phần quan trọng trong quá trình thích ứng của tế bào ung thư để phát triển di căn. Dựa trên các quan sát thực nghiệm, Lee và các đồng nghiệp cho rằng FAO là nguồn ATP chính trong các tế bào ác tính có nguồn gốc khác nhau. Ngoài ra, từ lâu người ta đã biết rằng ở các loại ung thư không phải là glycolytic như ung thư tuyến tiền liệt, ung thư hạch và ung thư biểu mô ống tụy, FAO là con đường nổi bật để sản xuất năng lượng.
Con đường tín hiệu gây ung thư thúc đẩy việc chuyển đổi trong quá trình chuyển hóa lipid. Ví dụ, c-MYC điều chỉnh quá trình tạo lipogenesis bằng cách tạo ra SREBP1. Chất hoạt hóa đồng phiên mã protein liên quan đến yes (YAP) thúc đẩy sự chuyển dịch trao đổi chất sang FAO trong di căn hạch bạch huyết. KRAS đột biến làm trung gian cho quá trình lập trình lại quá trình chuyển hóa lipid thông qua acyl-coenzyme A (CoA) synthetase thành viên họ chuỗi dài 3 (ACSL3) trong ung thư phổi. Trục PI3K/Akt/mTOR điều hòa tăng CD36 và SREBP1 và gây ra quá trình tạo lipogenesis. Đây chỉ là một số ví dụ về việc tế bào ung thư khai thác lipogenesis. Để biết thêm thông tin, vui lòng xem các bài đánh giá xuất sắc về chủ đề này.
Điều quan trọng là không chỉ các gen ung thư ảnh hưởng đến quá trình chuyển hóa lipid. Cách khác cũng có thể xảy ra, tức là quá trình chuyển hóa lipid có thể ảnh hưởng đến tín hiệu gây ung thư. Thành phần của màng tế bào (hồ sơ của các nhóm FA, hàm lượng sphingolipid và cholesterol, v.v.) có thể ảnh hưởng đáng kể đến các tầng tín hiệu. Ví dụ, mức độ bão hòa màng do sinh tổng hợp của một số enzym của axit béo thúc đẩy sự phân cụm EGFR và kích hoạt tín hiệu. Một ví dụ khác đến từ ung thư tuyến tiền liệt, trong đó các axit béo không bão hòa đa thay đổi hàm lượng phospholipid, do đó làm thay đổi hoạt động của PIP3/AKT.
Chế độ ăn nhiều lipid có liên quan chặt chẽ đến sự phát triển của ung thư. Ngoài ra, các nghiên cứu lâm sàng khác nhau cho thấy béo phì và nguy cơ ung thư có liên quan mật thiết với nhau. Các thí nghiệm khác nhau đã tiết lộ rằng môi trường giàu lipid lập trình lại các tế bào ác tính để hấp thụ và chuyển hóa FA để hỗ trợ sự phát triển ác tính. Việc tiêu thụ lipid ngày càng tăng thúc đẩy sự phát triển của ung thư và sự phát triển của di căn trong các mô hình ung thư vú, đại trực tràng và dạ dày ở chuột. Ví dụ, chế độ ăn nhiều chất béo làm tăng biểu hiện CD36 và gây di căn trong mô hình chuột ung thư dạ dày. Axit béo cao gây ra sự di chuyển và xâm lấn của các tế bào ung thư tuyến tụy và chuyển chúng sang quá trình trao đổi chất oxy hóa. Trong một nghiên cứu hấp dẫn của Lee và các đồng nghiệp, sự tăng trưởng khối u trong mô hình khối u chuột đột biến KRAS cao gấp hai lần ở những con chuột tiêu thụ nhiều chất béo so với nhóm đối chứng (được cho ăn bình thường). Tuy nhiên, sự tăng trưởng khối u chậm hơn ba lần ở nhóm tiêu thụ ít chất béo (nhưng cân bằng calo) so với nhóm đối chứng (chế độ ăn bình thường), cho thấy rằng sự tăng trưởng khối u phụ thuộc vào axit béo là nguồn năng lượng chính.
Có nhiều bằng chứng cho thấy các tế bào mỡ gần đó có thể gây ra quá trình lập trình lại quá trình chuyển hóa lipid trong tế bào ung thư. Việc đồng nuôi cấy các tế bào ung thư buồng trứng với các tế bào mỡ đã kích thích biểu hiện của CD36, một thụ thể FA, làm tăng tiềm năng di căn và tăng trưởng dị ghép. Hạ CD36 hoặc sử dụng các kháng thể đặc hiệu đã phá vỡ quá trình tái lập trình qua trung gian tế bào mỡ này. Nhìn chung, các tế bào mỡ liên quan đến ung thư ngụ ý nhiều cơ chế khác nhau để thúc đẩy sự phát triển của khối u. Có lẽ, đây có thể là một lời giải thích cho việc nhiều khối u thường di căn đến các mô giàu tế bào mỡ.
Tóm lại, những quan sát này nhấn mạnh quan điểm rằng việc tăng cường hấp thu lipid có thể lập trình lại quá trình trao đổi chất, do đó làm thay đổi các con đường tín hiệu và do đó thúc đẩy kiểu hình ác tính và sự phát triển của di căn.
3. Sự tương tác giữa các Con đường Sinh hóa Thúc đẩy Tính dẻo của Chuyển hóa
Hiện nay, người ta chấp nhận rộng rãi rằng việc tái cấu trúc chuyển hóa mang lại lợi thế chọn lọc cho tế bào ung thư không phải bằng cách đơn giản là làm mất quy định các con đường trao đổi chất của chúng mà là bằng cách tạo ra tính dẻo trao đổi chất, cho phép chúng chuyển đổi giữa các trạng thái khác nhau như một phần của quá trình thích nghi. Thật không may, các cơ chế phân tử chính xác làm cơ sở cho tính dẻo của quá trình trao đổi chất còn lâu mới được hiểu đầy đủ. Tuy nhiên, có rất nhiều báo cáo chứng minh rằng tính dẻo của quá trình trao đổi chất cung cấp cho tế bào ung thư năng lượng và các khối “xây dựng” cần thiết cho sự tăng sinh, xâm lấn, di căn và kháng lại hệ thống miễn dịch và liệu pháp điều trị.
Một trong những đặc điểm chính của việc tái cấu trúc và dẻo dai trao đổi chất là tính không đồng nhất về trao đổi chất của các tế bào ác tính, đây là đặc điểm của nhiều khối u. Đặc điểm này được minh họa rõ bằng sự tương tác giữa đường phân và OXPHOS.
Một ví dụ về sự tương tác như vậy là tế bào gốc ung thư (CSC). CSC là những tế bào có khả năng tự đổi mới và khởi phát khối u. Chúng chịu trách nhiệm cho sự tái phát ung thư và kháng thuốc. Trước đây người ta đã chấp nhận rằng CSC có kiểu hình đường phân nhiều hơn. Thật vậy, các CSC có nguồn gốc khác nhau, bao gồm ung thư vú, dạ dày và ung thư biểu mô tế bào gan, đã được báo cáo là biểu hiện cao các gen đường phân và tăng cường đường phân và mức OXPHOS thấp. Ngược lại, có một số báo cáo về CSC phụ thuộc OXPHOS từ u thần kinh đệm, bệnh bạch cầu, ung thư buồng trứng, gan và ung thư tuyến tụy.
Trên thực tế, sự xuất hiện đồng thời của cả CSCs glycolytic và OXPHOS đã được báo cáo đối với các loại khối u tuyến tụy, vú và các loại khối u khác.
Gần đây, người ta đã chỉ ra rằng có hai quần thể tế bào gốc trong u thần kinh đệm ở chuột đồng gen, một trong số đó là glycolytic, trong khi quần thể còn lại dựa vào OXPHOS tùy thuộc vào đặc điểm trao đổi chất của các tế bào khối u có nguồn gốc. Các tác giả báo cáo rằng cả hai kiểu hình đều độc lập và ổn định. Tuy nhiên, quần thể OXPHOS được chuyển sang đường phân trong điều kiện thiếu oxy hoặc chất ức chế trao đổi chất.
Do đó, một số tác giả đã đề xuất sự tồn tại của trạng thái chuyển hóa lai (glycolytic/OXPHOS), cho phép tế bào ác tính chuyển đổi sang chế độ trao đổi chất phù hợp nhất trong điều kiện cụ thể để thích nghi và tồn tại. Ví dụ, tế bào ung thư vú thường có tính không đồng nhất chuyển hóa cao, cho phép chúng xâm chiếm các hốc khác nhau. Mô phổi có mức oxy cao và di căn phổi bắt nguồn từ tế bào ung thư OXPHOS. Ngược lại, gan có mức oxy thấp nên di căn gan bắt nguồn từ tế bào ung thư glycolytic. Phù hợp với điều này, nghiên cứu cho thấy việc mất GPX2 làm tăng biểu hiện HIF1α và kiểu hình glycolytic trong khi giảm OXPHOS. Tuy nhiên, trong một cụm tế bào cụ thể, việc mất GPX2 đã gây ra kiểu hình lai với sự gia tăng cả hai dấu hiệu gen EMT/giống tế bào gốc được điều hòa bởi HIF1α và AMPK.
Nhìn chung, pyruvate có nguồn gốc từ đường phân là nguồn chính của TCA và OXPHOS. Tuy nhiên, không chỉ đường phân cung cấp năng lượng cho hô hấp. Tính dẻo của chuyển hóa bắt nguồn từ sự đa dạng của các chất chuyển hóa ty thể, có thể được sử dụng làm nhiên liệu năng lượng chính cho OXPHOS trong một số điều kiện nhất định (Hình 2). Các chất tương đương NADH và FADH2 có nguồn gốc từ quá trình chuyển đổi trao đổi chất của axit amin và quá trình oxy hóa β cũng có thể bị oxy hóa bởi các phức hợp chuỗi hô hấp ty thể.
Ví dụ, có thể có sự hợp tác giữa các tế bào ung thư glycolytic và OXPHOS hoặc giữa tế bào ung thư và tế bào mô đệm trong khối u. Trong trường hợp này, các tế bào phụ thuộc OXPHOS nhập khẩu lactate và chuyển đổi nó thành pyruvate, do đó cung cấp năng lượng cho TCA và OXPHOS. Hiện tượng này được gọi là hiệu ứng Warburg đảo ngược và đã được quan sát thấy, ví dụ, trong vi môi trường khối u, khi quá trình đường phân trong chất đệm liên kết với ung thư hỗ trợ trao đổi chất cho các tế bào ung thư liền kề.
Việc chuyển đổi serine thành glycine trong chu trình folate ty thể tạo ra một lượng đáng kể NADH2, có thể được OXPHOS sử dụng và cung cấp tính dẻo trao đổi chất cho tế bào ung thư vú.
Proline bị oxy hóa thành pyrroline-5-carboxylate bởi proline carboxylase, được liên kết với các phức hợp chuỗi hô hấp II và III. Phản ứng này tạo ra FADH2 và hỗ trợ quá trình hình thành khối u và sự phát triển của di căn phổi trong các mô hình chuột 4T1 và EMT6.5 nguyên vị.
Glutamine có nguồn gốc từ quá trình glutaminolysis được chuyển hóa thành α-KG, đi vào chu trình TCA và hỗ trợ OXPHOS. Ngoài ra, glutamate dehydrogenase (GDH), chịu trách nhiệm cho bước cuối cùng này, đi kèm với việc tạo ra NADH.
Do đó, để ngăn chặn sự tương tác phức tạp này giữa quá trình glycolysis và OXPHOS, mang lại cho các tế bào tân sinh những lợi thế về độ dẻo thích nghi, khả năng sống sót và tăng trưởng, chúng ta cần nhắm mục tiêu không chỉ các quá trình đơn lẻ mà còn cả mạng lưới đầy đủ và các chất trung gian phân tử quan trọng chi phối độ dẻo trao đổi chất này.
4. Các Con đường Tín hiệu Ung thư Điều chỉnh Sự Tái lập trình và Tính dẻo của Chuyển hóa
Khi nói về các chiến lược điều trị trong bối cảnh tái cấu trúc chuyển hóa ở ung thư, việc chỉ ức chế một số enzym chuyển hóa rõ ràng là không đủ để tạo ra hiệu quả điều trị bền vững. Để phát triển các phương pháp hiệu quả nhắm vào mục tiêu tái lập trình chuyển hóa, điều quan trọng nữa là phải xem xét các yếu tố thúc đẩy phân tử làm rối loạn và tính dẻo của chuyển hóa.
Các gen ung thư và gen ức chế khối u khác nhau có thể điều chỉnh các thay đổi chuyển hóa liên quan đến ung thư. Ví dụ điển hình nhất là các yếu tố phiên mã c-Myc và HIF1α, hai chất điều hòa chính của quá trình đường phân và các con đường trao đổi chất khác, có thể trực tiếp hoạt hóa hàng chục gen chuyển hóa.
Ngoài ra còn có một mạng lưới phức tạp các mạch điều hòa sau dịch mã của quá trình đường phân được trung gian bởi các gen ung thư chính như AKT, mục tiêu của rapamycin ở động vật có vú (mTOR), thụ thể yếu tố tăng trưởng biểu bì (EGFR), virus sarcoma chuột Kirsten (K-Ras), và các loại khác.
Là chất điều hòa chính của các con đường đồng hóa, mTOR thúc đẩy quá trình đường phân, một carbon và chuyển hóa lipid. Các tiểu đơn vị xúc tác của nó, mTORC1 và mTORC2, kích thích biểu hiện của GLUT1 và các chất trung gian quan trọng nhất của quá trình đường phân—HK2, PFK và PKM2—thông qua việc tăng cường điều hòa HIF1α và c-Myc; kích thích sinh tổng hợp các nucleotide purine và pyrimidine; kiểm soát sinh tổng hợp và quá trình oxy hóa β của axit béo bằng cách điều hòa SREBP và PPARγ; và trực tiếp phosphoryl hóa và kích hoạt ACLY . Sự tái cấu trúc chuyển hóa qua trung gian mTOR mang lại khả năng kháng thuốc hóa trị liệu.
Bằng nhiều cơ chế, bao gồm cả cách phụ thuộc hoặc không phụ thuộc mTOR, AKT dẫn đến việc kích hoạt SREBP, c-Myc, HIF1α và ATF4. Nó trực tiếp phosphoryl hóa HK2 và PFKB2 và điều hòa tăng biểu hiện GLUT1.
Trong các tế bào tuyến tụy tiền ác tính, KRAS đột biến thúc đẩy quá trình tái lập trình chuyển hóa để kích thích biểu hiện của HK2, LDHA, PDK, glutaminase 1 (GLS1), glutamate dehydrogenase 1 (GLUD1), và các transaminase (GOT1, GPT2, và PSAT1), làm cho các tế bào phụ thuộc vào glucose và glutamine. Ngoài ra, KRAS kích thích GLUT1 và dòng glucose đến PPP.
Cả EGFR và Her2 (thụ thể thuộc họ ErbB) đều là các tác nhân thúc đẩy quá trình tái lập trình chuyển hóa. EGFR và Her2 tăng cường đường phân trong ung thư vú âm tính ba. Sự hoạt hóa quá mức EGFR, do khuếch đại hoặc đột biến, gây ra sự tái cấu trúc chuyển hóa bằng cách kích hoạt con đường mTORC2/Akt/c-Myc. Có nhiều bằng chứng về việc điều hòa tăng cường quá trình tạo lipogenesis trong các tế bào ung thư đột biến EGFR kháng với TKI. Trong các tế bào ung thư phổi đột biến EGFR vẫn tồn tại theo chu kỳ, sự chuyển dịch của quá trình trao đổi chất theo hướng FAO đã được quan sát thấy khi điều trị bằng thuốc ức chế tyrosine kinase. Trong các tế bào ung thư vú biểu hiện quá mức Her2, thường có sự tăng cường chuyển hóa glutamine.
Trên thực tế, tình hình phức tạp hơn do có rất nhiều mi-RNA, ảnh hưởng đến cả mRNA của các enzym và chất điều hòa của chúng.
5. Các Hợp chất có Nguồn gốc Thực vật Nhắm Mục tiêu Nhiều Con đường Sinh hóa
5.1. Kaempferol
Kaempferol là một flavanol lần đầu tiên được chiết xuất từ thân rễ của Kaempferia galanga (Hình 6). Đây là một thành phần dinh dưỡng không độc hại, giá rẻ, khá phổ biến trong chế độ ăn uống hàng ngày. Kaempferol giảm thiểu các quá trình viêm và có thể làm giảm viêm xương khớp, viêm khớp dạng thấp, viêm đại tràng và loét dạ dày.
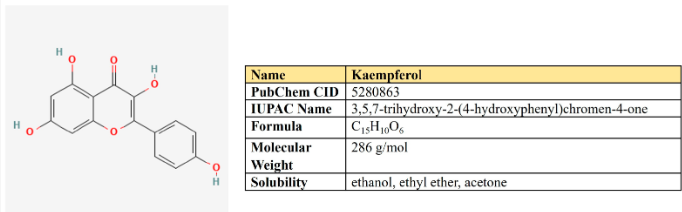
Hàng chục nghiên cứu đã chỉ ra hoạt tính chống ung thư của nó (Bảng 1). Kaempferol ức chế các con đường tín hiệu liên quan đến ung thư, ức chế sự tăng sinh, hình thành mạch và di chuyển, đồng thời đảo ngược khả năng kháng thuốc.
Một cơ chế khác của hoạt động chống ung thư của kaempferol có liên quan đến tác động tiêu cực của nó đối với các quá trình trao đổi chất. Yao và các đồng nghiệp đã chứng minh kaempferol qua trung gian EGFR ức chế sự hấp thu glucose và sản xuất lactate trong ung thư biểu mô thực quản. Trong trường hợp này, kaempferol ức chế EGFR và HK2 cả in vitro và in vivo.
Ngoài ra, kaempferol đã được chứng minh là ức chế quá trình đường phân trong ung thư ruột kết thông qua việc điều hòa tăng cường các micro-RNA đặc hiệu. Đầu tiên, kaempferol làm tăng biểu hiện của miR-339-5p, nhắm mục tiêu hnRNPA1 và PTBP1, từ đó tạo ra PKM2 khi ghép nối. Điều này dẫn đến giảm sản xuất axit lactic và ATP. Hơn nữa, kaempferol làm tăng biểu hiện của miR-326 nhắm mục tiêu trực tiếp PKM2, kèm theo khả năng đảo ngược kháng 5-FU.
Một cơ chế khác của quá trình ức chế đường phân qua trung gian kaempferol đã được đề xuất trong các tế bào u ác tính. Kaempferol ngăn cản sự liên kết của HK2 và VDAC1 trên ty thể thông qua con đường tín hiệu AKT/GSK-3β, ức chế sản xuất pyruvate và lactate và di căn.
Bên cạnh các enzym của quá trình đường phân, kaempferol được cho là ảnh hưởng tiêu cực đến các chất điều hòa phiên mã chính của nó: HIF1α và c-Myc. Kaempferol glycosides gây ra sự phân hủy HIF1α phụ thuộc ubiquitin-proteasome, ức chế tín hiệu thiếu oxy và biểu hiện của GLUT1 trong tế bào ung thư tuyến tụy. Trong các tế bào ung thư gan, kaempferol không làm thay đổi mức protein HIF1α nhưng thay đổi vị trí của nó bằng cách bất hoạt p44/42 MAPK . Ngoài ra, hai nhóm nhà nghiên cứu đã tiết lộ rằng kaempferol có thể liên kết G-quadruplex trong vùng promotor c-Myc, do đó ức chế sự biểu hiện của nó .
Ngoài đường phân, kaempferol có thể ức chế hô hấp trong tế bào Hela bằng cách ức chế phức hợp chuỗi hô hấp ty thể I. Điều này dẫn đến sự thất bại về năng lượng và gây ra quá trình tự thực bằng cách tăng AMPK .
Ngoài ra, Brusselman và các đồng nghiệp đã chỉ ra sự ức chế FASN và lipogenesis qua trung gian kaempferol trong các tế bào ung thư tuyến tiền liệt và ung thư vú.
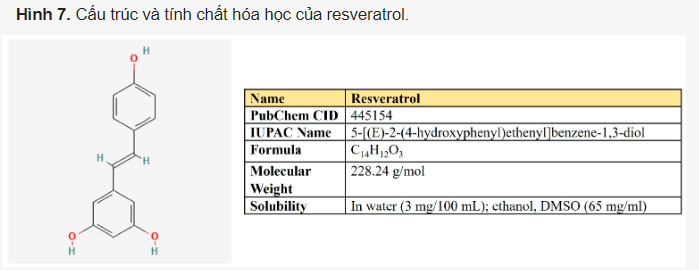
Resveratrol. Resveratrol là một 3,5,4′-trihydroxystilbene bao gồm hai vòng thơm được nối với nhau qua cầu methylene (Hình 7). Đây là một polyphenol trong chế độ ăn uống có hàm lượng đáng kể trong nho, rượu vang, đậu phộng và quả mọng. Nó cũng được tiêu thụ như một chất bổ sung chế độ ăn uống do các hoạt động chống ung thư, phòng ngừa ung thư, kháng vi-rút, kháng nấm, chống lão hóa và chống viêm.
Resveratrol có rất nhiều đặc tính chống ung thư liên quan đến tác động tiêu cực của nó đối với chu kỳ tế bào, sự hình thành mạch máu và các con đường tín hiệu tế bào, cũng như điều chế tích cực quá trình tự thực và apoptosis (Bảng 2). Các nghiên cứu khác nhau cũng liên kết các tác dụng chống ung thư qua trung gian resveratrol với việc kích hoạt p53 oncosupressor (được đánh giá trong).
Có nhiều báo cáo cho thấy resveratrol nhắm mục tiêu vào quá trình đường phân bằng nhiều cơ chế khác nhau trong các loại ung thư khác nhau. Thật vậy, resveratrol ức chế sự kích hoạt của EGFR, Akt và ERK1/2, dẫn đến ức chế quá trình đường phân qua trung gian HK2 ở NSCLC; điều hòa giảm HK2 trong ung thư biểu mô tế bào gan cả in vitro và in vivo. Nó ức chế quá trình đường phân trong tế bào ung thư tuyến tụy bằng cách nhắm mục tiêu miR-21.
Liên quan đến mô hình tạo mạch, resveratrol điều hòa giảm quá trình đường phân do VEGF gây ra trong tế bào nội mô tĩnh mạch rốn người (HUVEC), điều này có liên quan đến việc ức chế biểu hiện GLUT1, HK2, PFK1, PKM2 và sai vị trí PKM2.
Resveratrol không chỉ ảnh hưởng đến các enzym đường phân mà còn cả các chất điều hòa quan trọng của quá trình đường phân. Trong các tế bào ung thư buồng trứng, resveratrol ức chế quá trình đường phân, tăng sinh và di chuyển. Cơ chế phân tử đằng sau những tác động này liên quan đến việc kích hoạt AMPK và do đó ức chế mTOR. Trong các tế bào ung thư ruột kết và ung thư vú, resveratrol ức chế biểu hiện của c-Myc, VEGF và hTERT. Ở những con chuột mang khối u ung thư phổi Lewis, resveratrol ức chế lượng (18) F-FDG và đường phân làm giảm mức protein của HIF1α, Akt và mTOR. Các thí nghiệm gắn kết phân tử cho thấy resveratrol có thể là chất ức chế trực tiếp HIF1α; nó điều chỉnh giảm mức protein của nó trong các tế bào ung thư tuyến tụy.
Vanamala và các đồng nghiệp đã áp dụng phương pháp tiếp cận proteomic để tìm kiếm các protein bị resveratrol thay đổi trong tế bào ung thư ruột kết. Họ quan sát thấy G6PD và transketolase, hai enzym chính của con đường pentose phosphate (PPP), bị hợp chất này điều chỉnh giảm, liên kết resveratrol với việc điều chỉnh giảm PPP.
Một nhóm nghiên cứu khác đã chỉ ra rằng resveratrol ức chế c-Myc, tiêu thụ glucose và các enzym đường phân PK và LDH. Tuy nhiên, nó làm tăng citrate synthase, một trong những enzym của chu trình Krebs. Trong một nghiên cứu khác trên tế bào ung thư ruột kết, các tác giả cũng đã chỉ ra resveratrol làm giảm quá trình đường phân trong khi tăng quá trình oxy hóa glucose. Những quan sát này đi kèm với việc resveratrol điều chỉnh giảm PPP và lipogenesis. Tuy nhiên, các tác giả khác đã chỉ ra rằng cả quá trình đường phân và hô hấp đều bị resveratrol nhắm mục tiêu trong tế bào Hela, bao gồm một số protein OXPHOS chính.
Ngoài quá trình chuyển hóa glucose, resveratrol còn ức chế biểu hiện của chất nhập khẩu glutamine ASCT2 trong tế bào ung thư gan, làm tăng độ nhạy cảm với cisplatin. Ngoài ra, resveratrol có thể ức chế quá trình sinh tổng hợp axit béo de novo. Như đã đề cập ở trên, nó kích hoạt AMPK và điều chỉnh giảm mTOR trong tế bào ung thư vú, sau đó ức chế acetyl-CoA carboxylase α (ACACA) và axit béo synthase (FASN). Phù hợp với quan điểm này, resveratrol đã được chứng minh là làm giảm FASN trong ung thư vú biểu hiện quá mức Her2.
Tóm lại, có vẻ như tùy thuộc vào bối cảnh tế bào cụ thể, resveratrol có thể nhắm mục tiêu vào một loạt các con đường trao đổi chất trong tế bào ung thư, bao gồm đường phân, hô hấp, con đường pentose phosphate, sinh tổng hợp axit béo và hấp thu glutamine.
Quercetin. Quercetin là một hợp chất flavonoid (3,3′,4′,5,7-pentahydroxyflavone) được phân bố rộng rãi trong nhiều loại trái cây và rau củ khác nhau (Hình 8).
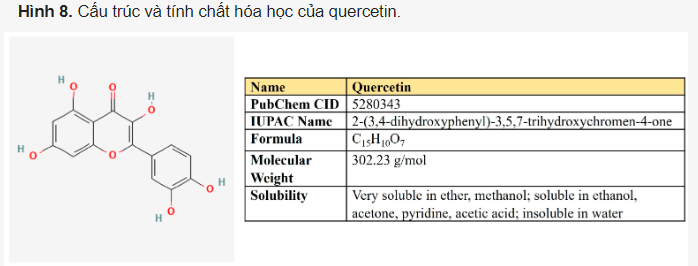
Có những báo cáo về việc quercetin ức chế một số con đường trao đổi chất quan trọng trong tế bào ung thư (Bảng 3). Trước hết, quercetin trực tiếp ức chế GLUT1 (Ki = 8 µM) trong các tế bào ung thư bạch cầu dòng tủy cấp tính (AML) HL-60.
Quercetin làm nhạy cảm các tế bào kháng erlotinib ung thư biểu mô tế bào vảy miệng bằng cách ức chế PKM2. Nó cũng ức chế GLUT1, HK2 và LDHA, cũng như Twist, N-cadherin, MMP-9 và MMP-13, làm giảm di chuyển, xâm lấn và tăng trưởng dị ghép. Trong ung thư vú, quercetin điều chỉnh giảm Akt, gây ra quá trình tự thực và ức chế sự hấp thu glucose, mức độ protein của PKM2, GLUT1, LDHA, MMP2, MMP9 và VEGF.
Hai nhóm nghiên cứu đã chứng minh rằng quercetin có thể liên kết các cấu trúc G-quadruplex trong chất khởi động c-Myc và ức chế sự biểu hiện của nó. Người ta cũng chỉ ra rằng quercetin làm giảm nhẹ con đường PI3K/Akt/mTOR và điều chỉnh giảm biểu hiện c-Myc trong bệnh ung thư hạch Burkitt. Hơn nữa, nó ức chế đáng kể mức protein của HIF1α và làm nhạy cảm các tế bào ung thư biểu mô tế bào gan và tuyến tụy với gemcitabine.
Quercetin không chỉ ức chế quá trình đường phân mà còn cả OXPHOS trong dòng tế bào u ác tính ở chuột. Hơn nữa, quercetin có thể giảm thiểu cả quá trình tổng hợp axit béo và quá trình oxy hóa β. Nó điều chỉnh giảm FASN trong tế bào HepG2 và ung thư vòm họng. Trong mô hình tế bào ung thư vú, Ruidas và các đồng nghiệp đã chỉ ra rằng quercetin điều chỉnh giảm mức biểu hiện của cả FASN và CPT1, cũng như cường độ oxy hóa β và sự phát triển của khối u in vivo. Hơn nữa, các phân tích gắn kết tính toán đã dự đoán sự liên kết của quercetin với CPT1, cho thấy tác dụng ức chế trực tiếp có thể có của hợp chất này đối với quá trình oxy hóa β.
(-)-Epigallocatechin-3-gallate (EGCG). EGCG là một hợp chất polyphenol (catechin), là este của epigallocatechin và axit gallic (Hình 9). Đây là catechin dồi dào nhất trong trà. Nhờ các đặc tính dược lý có lợi, bao gồm khả năng chống oxy hóa, bảo vệ tim và thần kinh, chống tiểu đường và giảm cholesterol, ngoài việc tiêu thụ hàng ngày dưới dạng trà, EGCG còn được sử dụng rộng rãi như một chất bổ sung chế độ ăn uống. Nghiên cứu đã chỉ ra rằng EGCG có thể ngăn ngừa lão hóa, rối loạn chức năng nhận thức và thậm chí là sự phát triển của ung thư.

Rất nhiều đặc tính chống ung thư làm cho EGCG trở thành ứng cử viên cho liệu pháp chống ung thư (Bảng 4). EGCG ức chế sự kích hoạt của tín hiệu c-Met và EGFR bằng cách thay đổi các bè lipid trên màng và ức chế EMT và sự xâm lấn bằng cách ức chế TGF-β1/Smad. Nó cũng có thể làm giảm các con đường qua trung gian STAT3, ERK NF-κB và Akt trong một số bệnh ung thư. Các nghiên cứu khác nhau đã chứng minh rằng EGCG làm nhạy cảm các tế bào khối u với các loại thuốc hóa trị thông thường như doxorubicin, cisplatin, 5-FU và tamoxifen cũng như có thể giúp giảm tác dụng phụ của chúng (được xem xét trong ).
Liên quan đến việc tái cấu trúc chuyển hóa liên quan đến ung thư, EGCG sở hữu đầy đủ các khả năng ức chế. Trước hết, EGCG ảnh hưởng đến quá trình đường phân theo nhiều cách, thể hiện tác dụng ức chế toàn cầu đối với năng lượng tế bào. Trong tế bào ung thư vú, nó gây ra quá trình tự thực và apoptosis, giảm mức lactate và ATP, ức chế mức mRNA và hoạt động của hexokinase (HK), phosphofructokinase (PFK) và lactic dehydrogenase (LDH); giảm tiêu thụ glucose, GLUT1 và HIF1α; ức chế sự tăng sinh và tăng trưởng dị ghép. Trong các tế bào ung thư lưỡi, EGCG ức chế sự hoạt hóa của EGFR, AKT và ERK1/2, làm giảm tiêu thụ glucose và sản xuất lactate, đồng thời làm giảm mức protein của HK2 và sự chuyển vị của nó đến màng ngoài ty thể.
Trong ung thư biểu mô tế bào gan, EGCG ức chế cả biểu hiện và hoạt động của PFK ở nồng độ 25–100 µM. Nó biến đổi cấu trúc oligomeric của PFK thành dạng không hoạt động, ức chế sự hấp thu glucose và sản xuất lactate, đồng thời gây ra apoptosis.
Sử dụng phương pháp tiếp cận chuyển hóa trong mô hình tế bào tuyến tụy, Lu và các đồng nghiệp đã tiết lộ sự nhiễu loạn do EGCG qua trung gian của mạng lưới chuyển hóa, điều hòa giảm tốc độ đường phân và sinh tổng hợp axit béo. Cuối cùng, trong tế bào ung thư ruột kết, EGCG can thiệp vào các bè lipid trên màng, làm giảm hoạt động của MCT1, qua đó làm trung gian cho quá trình xuất khẩu lactate — bước quan trọng hỗ trợ quá trình đường phân hiếu khí.
EGCG có thể ảnh hưởng tiêu cực đến chu trình Krebs biến đổi gen. Các khối u mang đột biến IDH sử dụng quá trình xử lý glutamine để tạo ra chất chuyển hóa oncometabolite, 2-hydroxyglutarate (2-HG). EGCG ở liều 5–20 µM ức chế cả IDH1 và GDH1/2, làm giảm sự tăng sinh và sản xuất 2-HG, làm cho tế bào ung thư đột biến IDH nhạy cảm với bức xạ.
Ngoài quá trình chuyển hóa glucose, một số nhà nghiên cứu đã phát hiện ra EGCG là chất ức chế trực tiếp glutamate dehydrogenase (GDH), loại enzyme này. Ngoài ra, EGCG là chất ức chế FASN trực tiếp. Puig và cộng sự đã so sánh tác dụng ức chế của EGCG và C75 đối với sự ức chế FASN trong tế bào ung thư vú. Trong khi mức độ ức chế tương tự đối với cả hai hợp chất, EGCG có tác dụng ức chế vừa phải đối với quá trình oxy hóa β của axit béo thông qua tác động tiêu cực đối với CPT1. Ngược lại, C75—một chất ức chế FASN thường được sử dụng—đã kích thích đáng kể CPT1. Cả phương pháp điều trị bằng EGCG và C75 đều dẫn đến giảm sự tăng sinh và mức protein của HER2, AKT và ERK1/2. Kết quả tương tự cũng thu được đối với ung thư phổi.
Trong tế bào ung thư biểu mô tế bào gan HepG2, EGCG đồng thời làm giảm mức protein FASN và ACC và giảm hoạt động của CPT1, có liên quan đến apoptosis.
Những dữ liệu này chứng minh tiềm năng cao của EGCG trong việc ức chế quá trình chuyển hóa trong ung thư.
5.2 Curcumin
Curcumin là một hợp chất polyphenol được chiết xuất từ thân rễ của nghệ (Curcuma longa L.) và là thành phần chính (Hình 10). Curcumin sở hữu một số đặc tính dược lý có lợi: chống oxy hóa, chống viêm, bảo vệ tim, gan và thần kinh, chống tiểu đường, chống loét, kháng khuẩn, v.v. Nó có hoạt tính chống lại ung thư vú, đại trực tràng, dạ dày, tuyến tiền liệt và ung thư phổi thông qua nhiều cơ chế phân tử khác nhau (Bảng 5). Tóm lại, curcumin ức chế một số con đường tín hiệu, bao gồm PI3K/AKT, ERK/MAPK, Wnt và NF-kβ; ức chế sự tăng sinh, di cư và xâm lấn; làm giảm tính gốc và gây ra quá trình tự thực, ferroptosis và apoptosis (được xem xét trong).

Một nhóm nghiên cứu đã chỉ ra rằng trong bốn dòng tế bào ung thư có nguồn gốc khác nhau, curcumin làm giảm sự hấp thu glucose, sản xuất lactate và mức protein của HIF1α, PKM2 và p70S6K – mục tiêu của mTOR. Hiệu ứng này đã bị bãi bỏ khi biểu hiện quá mức PKM2. Các nhà nghiên cứu khác đã nghiên cứu khả năng kháng hóa chất do tăng đường huyết qua trung gian curcumin trong các tế bào ung thư biểu mô tế bào gan. Curcumin làm giảm sự tồn tại của tế bào ung thư do glucose cao khi điều trị bằng doxorubicin và methotrexate, ức chế sự hấp thu glucose, sản xuất lactate, biểu hiện GLUT1/3, MCT1/4, HIF1α, mTOR, STAT3 và protein kháng đa thuốc MDR-1 .
Trong một số mô hình khối u ở chuột, curcumin đã được chứng minh là làm giảm hoạt động của ATP synthase, mức ATP và tỷ lệ ATP/AMP cả in vitro và in vivo. Nó cũng làm tăng ROS, gây ra autophagy và tiết lộ các hoạt động chống tạo mạch trong dị ghép B16 .
Trong tế bào MCF7, việc điều trị bằng curcumin đã được tăng cường gấp bốn lần sự hấp thu glucose, sản xuất lactate và hoạt động của HK. Điều này đi kèm với việc giảm đáng kể hô hấp 6 giờ sau điều trị cũng như ức chế sự phát triển của tế bào.
Tóm lại, có vẻ như tác dụng của curcumin đối với sự hấp thu glucose và hoạt động của các enzym đường phân phụ thuộc nhiều vào bối cảnh tế bào vì nó có thể khác biệt đối với các dòng tế bào có nguồn gốc khác nhau. Tuy nhiên, bất kể điều này, trong mọi trường hợp, curcumin đều ảnh hưởng tiêu cực đến sự phát triển của tế bào ác tính.
Curcumin làm giảm mạnh sự phát triển và di chuyển của các tế bào ung thư biểu mô tuyến thượng thận và cũng gây ra apoptosis trong các tế bào này. Mặc dù biểu hiện của một số gen đường phân được kích thích, cả đường phân và hô hấp đều bị ức chế đáng kể. Tuy nhiên, nó đi kèm với sự biểu hiện tăng cường của glutamic pyruvic transaminase (GPT), chất nhập khẩu glutamine SLC1A5 và glutaminase (GLS1), chỉ ra sự tái lập trình trao đổi chất theo hướng sử dụng glutamine. Hơn nữa, việc giảm nồng độ glutamine trong môi trường nuôi cấy đã làm tăng đáng kể đặc tính gây độc tế bào của curcumin. Những kết quả này cho thấy việc đồng thời nhắm mục tiêu vào quá trình chuyển hóa glutamine cùng với điều trị curcumin có thể là một chiến lược đầy hứa hẹn đối với ít nhất là ung thư biểu mô tuyến thượng thận.
Trong mô hình ung thư ruột kết, tình trạng kháng đa thuốc (P-gp) có liên quan chặt chẽ đến quá trình tổng hợp spermine và spermidine cũng như quá trình chuyển hóa glutamine. Curcumin ức chế những thay đổi chuyển hóa này, do đó làm giảm phản ứng chống oxy hóa và hoạt động vận chuyển P-gp và cuối cùng đảo ngược tình trạng kháng đa thuốc. Trong một nghiên cứu khác, curcumin ưu tiên nhắm mục tiêu vào CSCs ruột kết, ức chế quá trình chuyển hóa glutamine trong quần thể tế bào CD44+. Người ta đã chỉ ra rằng curcumin gây ra sự biểu hiện của miR-137, nhắm mục tiêu trực tiếp vào mRNA của glutaminase. Điều này làm suy yếu quá trình chuyển hóa glutamine và làm cho các tế bào ung thư đại trực tràng nhạy cảm với cisplatin.
Ngoài chuyển hóa glucose và glutamine, curcumin còn có tiềm năng cao trong việc nhắm mục tiêu chuyển hóa lipid, bao gồm các enzym tạo lipogenic FASN, ACC và ACLY, cũng như các chất điều hòa phiên mã của chúng, SREBP1. Do đó, curcumin điều chỉnh giảm cả mức độ biểu hiện và hoạt động của enzym FASN trong ung thư biểu mô tế bào gan (HCC) và ung thư vú. Trong mô hình HCC ở chuột, curcumin làm tăng đáng kể hoạt động của sorafenib, tăng số lượng tế bào T CD4+ và tế bào NK, điều chỉnh giảm p-PI3K/p-Akt, HIF1α, FASN, SREBP1 và CPT1a. Hơn nữa, mô hình máy tính đề xuất khả năng liên kết tiềm năng của curcumin với FASN, STAT3 và AKT.
Ngoài ra, rất nhiều dữ liệu về việc curcumin điều chỉnh giảm quá trình chuyển hóa lipid đến từ các nghiên cứu trên tế bào mỡ, tế bào gan và các tế bào và mô không phải khối u khác. Ví dụ, curcumin ức chế các gen liên quan đến quá trình sinh tổng hợp cholesterol, FASN, ACC, SREBP1 và PPARγ.
Trong một nghiên cứu khác, Yang và các đồng nghiệp đã phát triển các hạt nano mang curcumin, nhắm mục tiêu hiệu quả vào cả PKM2 và FASN và làm suy giảm quá trình trao đổi năng lượng trong các mô hình tế bào ung thư vú.
Arctigenin. Arctigenin (Arc) là một lignan (Hình 11) được tìm thấy trong cây ngưu bàng (Arctium lappa) có hoạt tính chống oxy hóa, chống viêm, kháng vi-rút và chống ung thư . Một phổ rộng các đặc tính chống ung thư đã được chứng minh cho hợp chất này trong các khối u khác nhau.
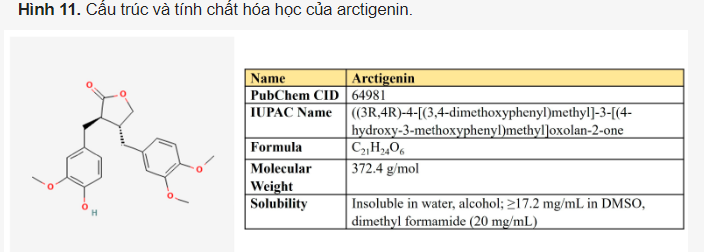
Trong các mô hình ung thư khác nhau, Arc ức chế các dòng tín hiệu qua trung gian EGFR và Her2, cũng như các con đường phụ thuộc AKT/mTOR và STAT3/β-catenin. Nó gây ra ngừng chu kỳ tế bào, apoptosis và autophagy, ức chế di căn và tạo mạch, đồng thời làm cho các tế bào ác tính nhạy cảm với hóa trị liệu.
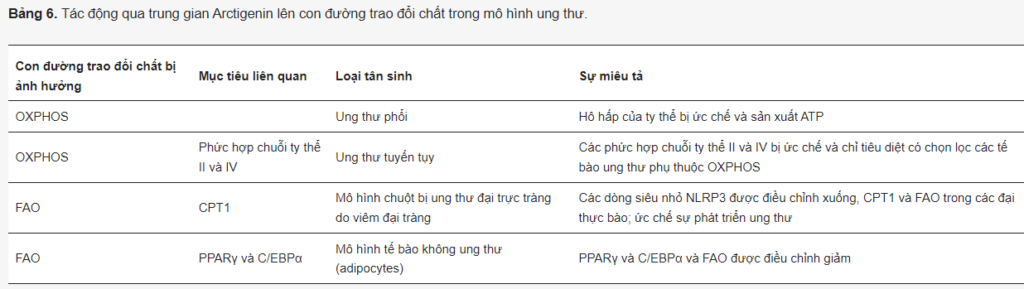
Trái ngược với các hợp chất khác được xem xét, Arc không ức chế quá trình đường phân. Tuy nhiên, nó ức chế OXPHOS và chuyển hóa lipid (Bảng 6). Trong mô hình tế bào ung thư phổi, Arc ức chế hô hấp ty thể và sản xuất ATP. Nó cũng hiệp đồng với 2-DG để gây ra cái chết tế bào ưu tiên của ung thư chứ không phải tế bào bình thường. Tác dụng ức chế của Arc đối với hô hấp cũng đã được chứng minh đối với ung thư tuyến tụy. Brecht và các đồng nghiệp đã tiết lộ rằng, về mặt cơ học, Arc nhắm mục tiêu các phức hợp chuỗi ty thể II và IV và chỉ tiêu diệt chọn lọc các tế bào ung thư tuyến tụy phụ thuộc OXPHOS. Cần lưu ý rằng Arc nhắm vào phức hợp chuỗi hô hấp I trong cơ xương, gây ra kích hoạt AMPK và có tác dụng có lợi đối với các rối loạn chuyển hóa trong mô hình chuột béo phì.
Brecht và cộng sự đã chỉ ra rằng việc Arc nhắm mục tiêu vào các tế bào ung thư tuyến tụy phụ thuộc OXPHOS đi kèm với việc gây ra stress ER (tăng GRP78, CHOP và ATF4). Tuy nhiên, trong điều kiện thiếu glucose, Arc có thể ức chế phản ứng protein chưa gấp bằng cách giảm GRP78, GRP94 và ATF4.
Các loại liệu pháp chống ung thư khác nhau gây ra stress ER. Các chất trung gian phản ứng protein chưa gấp, chẳng hạn như GRP78, PERC, CHOP, ATF4 và các chất khác, chịu trách nhiệm giảm thiểu stress ER, dẫn đến sự tồn tại của các tế bào ác tính. Do đó, việc nhắm mục tiêu vào các protein gây stress ER hiện được coi là một phương pháp chống ung thư.
Nhìn chung, những dữ liệu này cho thấy tiềm năng sử dụng Arc kết hợp với bất kỳ hợp chất nào nhắm mục tiêu vào quá trình đường phân và hấp thu glucose. Sự thiếu hụt glucose và ức chế quá trình đường phân gây ra cả stress ER và hô hấp như một cách thích nghi để ngăn ngừa thiếu hụt năng lượng. Vì Arc ức chế hô hấp và stress ER khi thiếu glucose, nó có thể mang lại hiệu quả hiệp đồng với các chất ức chế hấp thu glucose và đường phân.
Arc có thể ảnh hưởng không chỉ đến hô hấp mà còn có hoạt tính ức chế đối với quá trình chuyển hóa axit béo. NLRP3 inflammasome đóng một vai trò quan trọng trong sự phát triển của viêm đại tràng và ung thư đại trực tràng. Qiao và các đồng nghiệp đã thiết lập một mô hình chuột bị ung thư đại trực tràng do cảm ứng và nghiên cứu tác dụng điều trị tiềm năng của Arc. Họ quan sát thấy Arc làm giảm hoạt động của NLRP3 inflammasome và quá trình oxy hóa β của axit béo trong đại thực bào. Về mặt cơ chế, các xét nghiệm chuyển hóa và chuyển hóa cho thấy Arc làm giảm FAO, ức chế mức độ biểu hiện và hoạt động của enzym CPT1. Do đó, Arc ngăn ngừa sự tiến triển của viêm đại tràng và bảo vệ chống lại quá trình gây ung thư ruột kết.
Trong một nghiên cứu khác, Arc đã được chứng minh là làm giảm thụ thể gamma hoạt hóa peroxisome proliferator (PPARγ) và protein alpha liên kết CCAAT/tăng cường (C/EBPα) trong các tế bào mỡ biệt hóa, điều này cũng ngụ ý hoạt tính ức chế của tự nhiên này. hợp chất trên chuyển hóa axit béo.
Một số nghiên cứu đã báo cáo tác động tiêu cực của Arc đối với con đường Akt/mTOR, làm tăng các quá trình đồng hóa trong tế bào ác tính. Arc làm giảm phosphoryl hóa Akt và mTOR và gây ra autophagy ở ung thư tuyến tiền liệt, ung thư vú, ung thư gan và u nguyên bào thần kinh đệm.
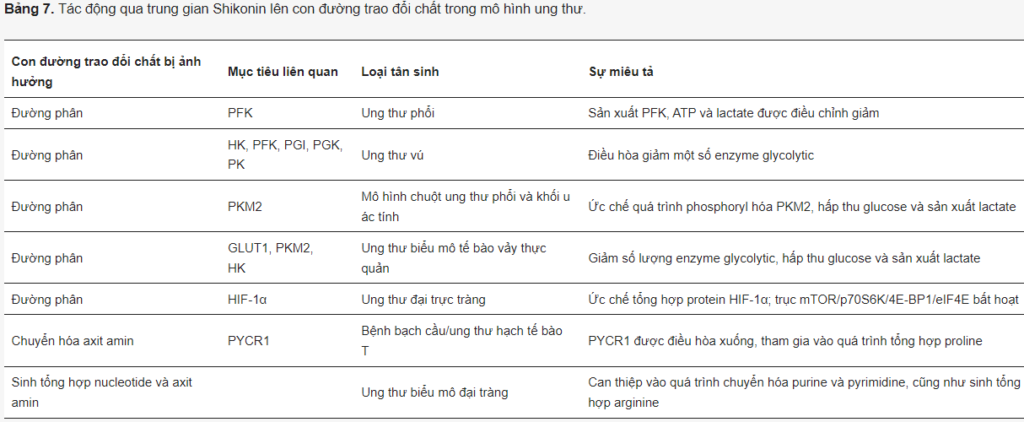
Trong tế bào ác tính, shikonin ức chế tín hiệu ERK và β-catenin, điều hòa tăng p21 và làm ngừng tế bào ở pha G2/M, nhắm mục tiêu các phosphatase chu kỳ phân chia tế bào 25 (Cdc25), gây apoptosis thông qua kích hoạt FOXO3a/EGR1/SIRT1. Ngoài ra, nó còn có tác động tiêu cực đến quá trình trao đổi chất của ung thư (Bảng 7).
Trong tế bào ung thư phổi, shikonin ức chế PFKB ở cả cấp độ mRNA và protein. Nó làm giảm sự tăng sinh, di chuyển, xâm lấn, hấp thu glucose, sản xuất ATP và lactate ở liều 10–50 µM, cũng như làm tăng số lượng tế bào apoptosis.
Chen và cộng sự. đã nghiên cứu hoạt tính ức chế của shikonin trên một tập hợp các enzym đường phân trong dịch chiết tế bào có nguồn gốc từ tế bào ung thư vú MCF7. Các tác giả đã thực hiện ủ tế bào với shikonin trong 1 giờ sau đó đo hoạt tính enzyme. Điều thú vị là nồng độ shikonin bên trong tế bào cao hơn nồng độ ngoại bào của nó, điều đó có nghĩa là tế bào có thể tích tụ shikonin. Giá trị IC50 đối với sự ức chế enzym lần lượt là 9,7, 17,2, 96,8, 89,5 và 12,2 µM đối với HK, PFK-1, PGI, PGK và PK.
Tuy nhiên, trước hết, shikonin được biết đến như một chất ức chế PKM2. Shikonin đã được chứng minh là ức chế đáng kể sự phát triển của tế bào khối u in vitro và in vivo trong mô hình chuột nguyên vị – ung thư biểu mô Lewis và u ác tính B16. Trong các hệ thống này, shikonin đã cải thiện quá trình phosphoryl hóa PKM2 nhưng không làm thay đổi mức protein của nó. Shikonin làm giảm sự hấp thu glucose và sản xuất lactate gây ra apoptosis ở liều 10–20 µM.
Trong dị ghép có nguồn gốc từ bệnh nhân ung thư biểu mô tế bào vảy thực quản, shikonin đã điều chỉnh giảm p-PKM2, HK, GLUT1 và p-STAT3, hấp thu glucose và sản xuất lactate, kèm theo sự ức chế tăng trưởng khối u.
Hóa trị liệu dựa trên Cisplatin thường được khắc phục trong ung thư bàng quang. Tuy nhiên, shikonin đã được báo cáo là có thể khắc phục tình trạng kháng cisplatin, phụ thuộc vào tác dụng ức chế đối với PKM2 và quá trình đường phân hiếu khí.
Ngoài ra, shikonin ngăn ngừa quá trình tạo dimer và tetramer của PKM2 trong đại thực bào, làm giảm viêm đại tràng ở chuột.
Đáng chú ý, shikonin ức chế sự tăng trưởng của tế bào ung thư đường mật và gây ra apoptosis lên đến 70% ở liều 0,5–1,5 µM, có nghĩa là các loại khối u này có thể cực kỳ nhạy cảm với hợp chất này. Trong ung thư đại trực tràng, shikonin ức chế quá trình tổng hợp protein HIF-1α mà không ảnh hưởng đến biểu hiện của mRNA HIF-1α hoặc làm suy giảm protein HIF-1α, dẫn đến bất hoạt mTOR/p70S6K/4E-BP1/eIF4E.
Ngoài chuyển hóa năng lượng, shikonin được báo cáo là ức chế enzym quan trọng của quá trình chuyển hóa axit amin – PYCR1, trong bệnh bạch cầu/u lympho tế bào T, kết hợp với ALDH18A1 tạo ra proline từ glutamate. Điều này có liên quan đến việc điều hòa tăng cường quá trình tự thực và apoptosis. Đáng chú ý là PYCR1, cùng với ALDH18A1, là hai enzym được biểu hiện quá mức nhiều nhất trong số 19 loại khối u và các gen được điều hòa tăng cao nhất trong ung thư biểu mô tế bào gan.
Chen và các đồng nghiệp cũng đã chỉ ra rằng chỉ cần 1 µM shikonin là đủ để điều chỉnh giảm hơn 50% tế bào ung thư biểu mô ruột kết. Các tác giả đã kết hợp dữ liệu phiên mã và dữ liệu chuyển hóa và chứng minh rằng quá trình chuyển hóa purine và pyrimidine, cũng như quá trình sinh tổng hợp arginine và chuyển hóa các axit amin khác, bị ảnh hưởng bởi sự can thiệp của shikonin. Hơn nữa, dNTP bổ sung và arginine đã giải cứu tình trạng gây độc tế bào do shikonin gây ra. Những kết quả này cho thấy tác động tiêu cực toàn cầu của shikonin đối với các mạng lưới trao đổi chất của ung thư.

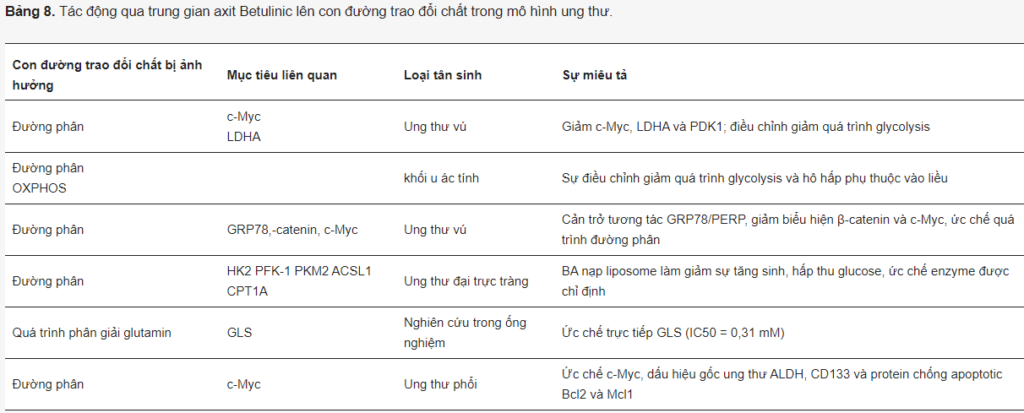
BA cản trở cường độ đường phân, hấp thu glucose và sản xuất lactate, đồng thời ức chế c-Myc, LDHA và PDK1 trong tế bào ung thư vú (Bảng 8). Trong tế bào u ác tính ở người, BA làm giảm cả quá trình đường phân và hô hấp theo cách phụ thuộc liều lượng. Đối với ung thư vú, BA cũng ức chế quá trình đường phân và sự phát triển của di căn in vivo. Về mặt cơ chế, nó ngăn chặn sự tương tác của protein 78 (GRP78) được điều hòa bằng glucose với cảm biến căng thẳng lưới nội chất (PERP), dẫn đến ức chế biểu hiện β-catenin và c-Myc, cũng như đường phân qua trung gian c-Myc. Trong tế bào ung thư phổi, BA cũng điều hòa giảm c-Myc và các dấu hiệu tế bào gốc ung thư CD133 và ALDH. Trong nghiên cứu khác, BA ức chế aldolase, enolase, LDHA và PKM2 trong ung thư đại trực tràng.
Bên cạnh đường phân, BA được chứng minh là điều hòa tiêu cực quá trình chuyển hóa glutamine và lipid. Trong các nghiên cứu gần đây, liposome chứa BA nhắm mục tiêu hiệu quả vào quá trình đường phân, glutaminolysis và chuyển hóa axit béo. Trong nghiên cứu của Wang và các đồng nghiệp, liposome như vậy đã ức chế sự tăng sinh và hấp thu glucose, giảm các enzym đường phân HK2, PFK-1, PEP và PKM2 cũng như một enzym quan trọng trong quá trình sinh tổng hợp axit béo—ACSL1 và enzym FAO giới hạn tốc độ—CPT1a.
Người ta cũng chỉ ra rằng BA trực tiếp ức chế glutaminase với IC50 là 0,31 mM, mặc dù nồng độ này có vẻ khá cao.
Mặc dù chưa có thông tin nào về tế bào ác tính, nhưng một vài bài báo mô tả rằng BA làm giảm sự tích tụ lipid trong tế bào mỡ bằng cách điều chỉnh PPARγ. Tuy nhiên, tác động tiềm tàng của BA đối với quá trình chuyển hóa lipid trong ung thư vẫn còn phải được giải quyết.
5.5 Cucurbitacins
Cucurbitacins là một nhóm các hợp chất triterpenoid tetracyclic được sản xuất bởi các thành viên của họ Bầu bí (Cucurbitaceae), bao gồm dưa chuột, bí ngô, dưa và dưa hấu (Hình 14). Các hợp chất này thu hút sự chú ý bởi đặc tính chống ung thư, chống viêm, kháng vi-rút, kháng khuẩn, tăng đường huyết, chống oxy hóa và bảo vệ gan ở người. Cucurbitacin B (CucB) đã được sử dụng trong y học Trung Quốc dưới dạng viên nén.
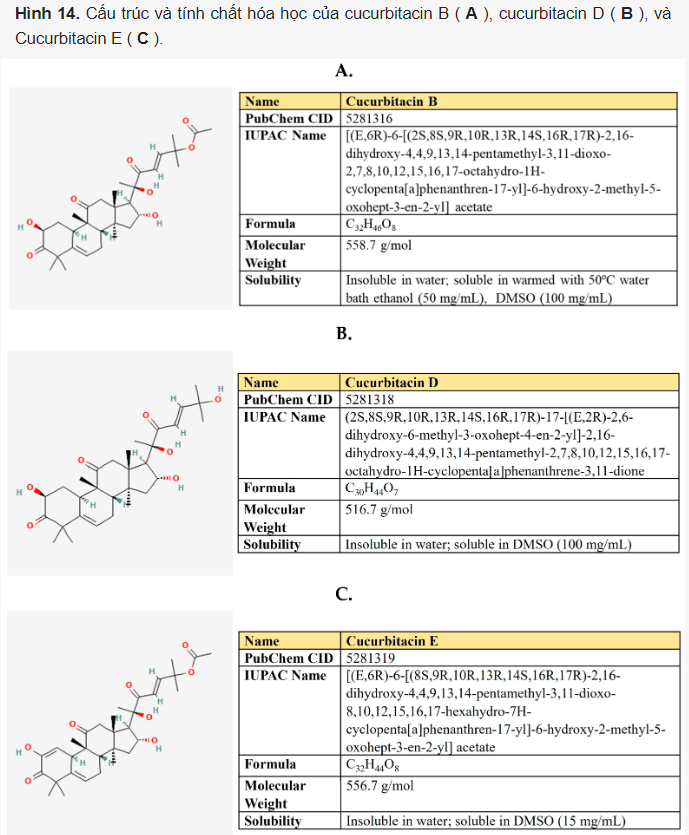
Theo dữ liệu từ các tài liệu nghiên cứu, cucurbitacin mang lại hiệu quả chống ung thư mạnh mẽ đối với các loại ung thư khác nhau – vú, phổi, tuyến tiền liệt, tuyến tụy, dạ dày, v.v., ảnh hưởng tiêu cực đến các con đường tín hiệu Jack/STAT, NFκB, PI3K/Akt/mTOR, MAPK/ERK và Wnt/β-catenin. Một số nghiên cứu đã chỉ ra rằng cucurbitacin làm tăng khả năng nhạy cảm của các tế bào ung thư với các liệu pháp chống ung thư, bao gồm doxorubicin, paclitaxel, dodetaxel, cisplatin, irinotecan, gemcitabine và methotrexate (được tóm tắt trong).
Trong ung thư tuyến tiền liệt, cucurbitacin D ở nồng độ dưới micromolar làm giảm sự hấp thu glucose và sản xuất lactate, đồng thời giảm AKT, GLUT1 và mức protein c-Myc (Bảng 9). Nó làm trung gian cho quá trình ngừng chu kỳ tế bào G2/M và apoptosis và làm suy giảm sự phát triển của dị ghép. Trong một nghiên cứu khác về ung thư tuyến tiền liệt, cucurbitacin B cũng làm giảm sự tăng trưởng của dị ghép và gây ra apoptosis, phụ thuộc vào sự điều hòa giảm phosphoryl hóa ACLY qua trung gian CucB. Ji và các đồng nghiệp đã điều tra tác động của CucB đối với sự trao đổi chất khối u của ung thư biểu mô tế bào gan biểu hiện quá mức c-Met/AKT ở chuột. Họ quan sát thấy hoạt tính ức chế của CucB đối với một số mạng lưới trao đổi chất, bao gồm cả quá trình tạo lipogenesis de novo. CucB ức chế hoạt động của AKT và mTOR, các enzym đường phân HK2 và PKM2, điều hòa giảm SPEBP1 và hai mục tiêu phiên mã chính của nó là FASN và ACC.
Trong tế bào ung thư vú, CucB ức chế biểu hiện của telomerase (hTERT) và c-Myc. Điều thú vị là, trong một nghiên cứu khác về ung thư vú, các tác giả đã báo cáo rằng CucB làm tăng đáng kể mức DNMT1 và gây ra quá trình methyl hóa rộng rãi các vùng promoter của gen c-Myc, cyclin D1 và survivin, dẫn đến việc điều hòa giảm các gen gây ung thư này và ức chế sự phát triển của tế bào ung thư.

Việc điều trị các dòng tế bào ung thư xương bằng cucurbitacin I đã ức chế tín hiệu STAT3 và làm giảm biểu hiện của cyclin D1, Mcl-1, c-Myc và khả năng sống sót.
Jiang và các đồng nghiệp đã nghiên cứu ảnh hưởng của CucE đối với các đặc tính của tế bào gốc ung thư thanh quản (LCSC). Các tác giả đã chỉ ra rằng điều trị bằng CucE làm giảm đáng kể khả năng xâm lấn của LCSC và ức chế sự phát triển của dị ghép hiệu quả hơn doxorubicin một phần tư, cũng như tăng cường độc tính do doxorubicin gây ra. Hơn nữa, CucE làm giảm mức protein của c-Myc và các protein kháng đa thuốc ABCG2 và P-gp, được gọi là dấu hiệu LCSC. Ngoài c-Myc, CucB còn được chứng minh là ức chế quá trình tổng hợp protein HIF-1α mà không có bất kỳ tác động nào đến mức độ phiên mã trong tế bào ung thư cổ tử cung cả in vitro và in vivo. Hiệu ứng này đi kèm với việc bất hoạt ERK1/2, mTOR và các chất hiệu ứng hạ lưu của nó: ribosomal protein S6 kinase (p70S6K) và eukaryotic initiation factor 4E-binding protein-1 (4E-BP1).
Trong tế bào ung thư biểu mô phổi không phải tế bào nhỏ, cucurbitacin B gây ra sự thoái hóa lysosomal của EGFR ở liều 0,01–0,1 µM và ức chế mạnh sự phát triển của dị ghép. Hiệu ứng này có giá trị chuyển đổi cao vì EGFR đột biến mang lại sự tăng sinh cấu thành cho tế bào ung thư và kháng lại các liệu pháp chống EGFR tiêu chuẩn, tạo ra các vấn đề cho liệu pháp điều trị ung thư phổi. Bằng cách hoạt động ở mức độ thoái hóa lysosomal của EGFR, CucB có thể nhắm mục tiêu hiệu quả các tế bào ung thư mang EGFR kiểu hoang dã hoặc đột biến.
5.6 Ginsenosides
Ginsenosides là một nhóm các hợp chất chủ yếu chịu trách nhiệm về các đặc tính y học của nhân sâm – một trong những loại cây phổ biến nhất của y học cổ truyền. Về cấu trúc hóa học, ginsenosides là saponin steroid có bản chất triterpenoid dammarane, có cấu hình giống steroid bốn vòng với các phần đường liên hợp (Hình 15). Khoảng 100 ginsenosides khác nhau đã được phân lập từ rễ, thân và lá của các loài Panax, và đáng chú ý là 10 trong số chúng được đặc trưng và sở hữu các đặc tính dược lý nổi tiếng.
Trong ung thư thực quản, ginsenoside Rh4 ở liều 20–60 µM có thể làm giảm mức protein của GLUT1, HK2, PFKL, PKM2, LDHA, p-Akt và p-mTOR. Quan sát này có liên quan đến việc giảm mức độ đường phân và hô hấp, sản xuất ATP và lactate. Trong NSCLC, ginsenoside Rh2 nhắm mục tiêu trục STAT3/c-Myc, dẫn đến ức chế GLUT1, PKM2 và LDHA và có liên quan đến việc ức chế EMT và gây ra apoptosis.
Một loại ginsenoside khác, hợp chất K (CK), làm suy yếu tín hiệu AKT/mTOR/c-Myc trong ung thư biểu mô tế bào gan, cũng dẫn đến ức chế HK2 và PKM2 và gây ra apoptosis.
Bên cạnh trục AKT/mTOR/c-Myc, ginsenosides ức chế các con đường phụ thuộc HIF1α. Trong các tế bào ung thư gan thiếu oxy và các mô hình dị ghép, ginsenoside CK ở liều 20–60 µM điều chỉnh giảm mức protein của HIF1α, đường phân, GLUT1, các enzym đường phân HK2 và LDHA, cũng như PDK1. Trong ung thư buồng trứng, ginsenoside Rh3 điều hòa tăng miR-519a-5p, lần lượt nhắm mục tiêu HIF1α. Nó ức chế quá trình methyl hóa DNA do DNMT3A làm trung gian trong vùng promoter của miR-519a-5p.
Ginsenoside Rk1 ức chế tín hiệu ERK/c-Myc, điều hòa giảm glutaminase GLS1 và giảm sản xuất glutathione, kích thích ROS và apoptosis trong ung thư biểu mô tế bào gan.
Một báo cáo khác về tác động của ginsenoside đối với quá trình chuyển hóa glutamine đến từ ung thư vú âm tính ba. Zhang và đồng tác giả đã báo cáo rằng việc điều trị bằng ginsenoside CK làm giảm chất vận chuyển glutamine ASCT2, glutaminase GLS1, glutamate dehydrogenase GLUD1 (GDH) và chất điều hòa phiên mã c-Myc của chúng ở cả mức mRNA và protein. Phù hợp với việc ức chế hấp thu glutamine và glutaminolysis, CK làm tăng mức glutamine và giảm glutamate, proline, aspartate và asparagine. Hơn nữa, CK ức chế sản xuất ATP và glutathione, do đó làm tăng ROS.
Ngoài ra còn có bằng chứng về tác động tiêu cực tiềm tàng qua trung gian ginsenosides đối với quá trình chuyển hóa lipid. Do đó, ginsenosides Rb1, Rg1, Rg3 và CK làm giảm cholesterol nội bào và thúc đẩy quá trình vận chuyển cholesterol ra khỏi tế bào u nguyên bào thần kinh đệm, cản trở sự phân bố các bè lipid trên màng và đảo ngược khả năng kháng temozolomide. Liên quan đến chuyển hóa lipid trong các mô hình không ung thư, ginsenosides được biết là ức chế FASN và SREBP1.
Cho đến nay, chúng tôi đã thảo luận về một số hợp chất tự nhiên có nguồn gốc thực vật ảnh hưởng tiêu cực đến các con đường trao đổi chất khác nhau trong các bệnh ác tính. Sơ đồ tóm tắt về cách các hợp chất này tác động đến các con đường sinh hóa khác nhau được thể hiện trong Hình 16.
6. Sinh khả dụng và Tính an toàn của các Hợp chất được Đánh giá
Mặc dù các hợp chất tự nhiên có đặc tính chống ung thư nổi bật, nhưng việc chuyển chúng sang nghiên cứu và trị liệu trên người thường bị hạn chế do sinh khả dụng thấp. Sinh khả dụng là phần chất được cơ thể hấp thụ và có sẵn để tham gia vào các hoạt động sinh lý. Nó có nguồn gốc từ sự hấp thụ, phân phối, chuyển hóa và bài tiết (ADME). Độ hòa tan trong nước thấp là một trong những thách thức chính. Sự chuyển hóa của các hợp chất chủ yếu bao gồm quá trình chuyển hóa ở ruột (bởi cả vi sinh vật và tế bào ruột) và gan. Nói chung, có các enzym chuyển hóa thuốc giai đoạn I và giai đoạn II, bao gồm cytochrome P (CYPs) và UDP-glucuronosyltransferases (UGTs).
Thông thường, sinh khả dụng và hoạt tính sinh học của một hợp chất nhất định có thể được cải thiện bằng cách sử dụng chất tăng cường sinh học hoặc hệ thống mang thuốc, có thể bao gồm liposome, hạt nano bạc hoặc silica, PLGA (axit poly-lactic-co-glycolic), PLA (poly(D,L-axit lactic)) các hạt nano, micelles polyme, hạt nano chitosan và các loại khác. Ví dụ, các hệ thống phân phối thuốc tự vi nhũ hóa (SMEDDS) thường được phát triển, là hỗn hợp đẳng hướng của dầu, chất hoạt động bề mặt hoặc (cách khác) chất hoạt động bề mặt và chất đồng dung môi. Việc áp dụng SMEDDS đã cải thiện đáng kể độ ổn định, hiệu quả, nồng độ thuốc tối đa (Cmax) và giá trị diện tích dưới đường cong (AUC) của curcumin, quercetin và resveratrol.
Ít nhất một thử nghiệm lâm sàng với kết quả đáng kể ở giai đoạn III có đối chứng là cần thiết để Cục Quản lý Thực phẩm và Dược phẩm (FDA) và Cơ quan Dược phẩm Châu Âu (EMA) đưa hợp chất vào sử dụng lâm sàng. Ngay cả những chất được tiêu thụ rộng rãi cũng có thể gây ra mức độ độc hại nguy hiểm khi chúng được tiêu thụ với liều lượng tăng lên. Ví dụ, những người tiêu thụ quá nhiều trà xanh hoặc chiết xuất của nó dưới dạng thực phẩm bổ sung đã bị nhiễm độc gan nghiêm trọng.
Do đó, để chuyển thành phương pháp điều trị, tất cả các hợp chất tự nhiên nên được nghiên cứu trong một loạt các thử nghiệm tiền lâm sàng và lâm sàng về độ an toàn của chúng. Dưới đây, chúng tôi thảo luận ngắn gọn về sinh khả dụng và tính an toàn của các hợp chất tự nhiên được đánh giá.
Kaempferol. Kaempferol có độ tan trong nước, sinh khả dụng và khả năng hấp thụ thấp. Vì vậy, việc hấp thụ 15 mg kaempferol ở người dẫn đến nồng độ trong huyết tương là 58 nM. Tuy nhiên, các thí nghiệm trên chuột cho thấy thời gian bán hủy của kaempferol cực kỳ ngắn, khoảng 4 phút.
Để giải quyết vấn đề sinh khả dụng kém, các hạt nano mang kaempferol đã được phát triển, có hoạt tính chống ung thư in vitro và in vivo đối với ung thư biểu mô tế bào gan.
Quercetin. Mặc dù một số nghiên cứu đã chỉ ra mối liên hệ trực tiếp giữa việc ăn quercetin và hoạt tính sinh học có lợi của nó ở người (được tóm tắt trong), nhưng sinh khả dụng của nó thường bị hạn chế bởi độ hòa tan trong nước kém và sự ổn định hóa học trong thực phẩm và ruột của con người. Do đó, các công nghệ đóng gói khác nhau đã được áp dụng để cải thiện sinh khả dụng của quercetin (được xem xét trong). Ví dụ, gần đây, các hạt nano PLGA chứa quercetin đã được phát triển, nhắm mục tiêu hiệu quả vào ung thư biểu mô tuyến vú ở chuột.
Có bốn nghiên cứu lâm sàng về tính an toàn của quercetin (được xem xét trong). Trong tất cả các nghiên cứu, quercetin được ăn dưới dạng aglycone; một liều duy nhất dao động từ 150 đến 5000 mg. Chỉ có một nghiên cứu báo cáo sự gia tăng nhỏ TNF-α, trong khi các nghiên cứu khác không báo cáo tác dụng phụ đáng kể nào. Do đó, quercetin có trạng thái của FDA (Cục Quản lý Thực phẩm và Dược phẩm) “Được công nhận là An toàn (GRAS)” như một chất bổ sung thực phẩm lên đến 500 mg mỗi khẩu phần.
EGCG. Mặc dù có bản chất ưa nước nhưng EGCG có sinh khả dụng đường uống thấp, thấp nhất trong số các catechin khác. Nó có độ ổn định cực thấp sau khi tiêu hóa, với <10% có sẵn để hấp thụ. Điều thú vị là việc sử dụng trà xanh với sữa, axit ascorbic hoặc nước trái cây sẽ cải thiện đáng kể sinh khả dụng của EGCG. Bên cạnh độ ổn định thấp, quá trình trao đổi chất và biến đổi sinh học xảy ra ở miệng, ruột và gan, làm giảm hơn nữa sinh khả dụng. Mặc dù vậy, một số nghiên cứu đã chỉ ra rằng việc uống chiết xuất trà xanh đã khử caffein trong viên nang—Polyphenon E, Teavigo® và FontUp®, đại diện cho hỗn hợp catechin khi bụng đói sau một đêm nhịn ăn có thể tăng cường sinh khả dụng và dẫn đến lượng đáng kể của nó. nồng độ trong máu.
Tuy nhiên, các hệ thống phân phối nano cải thiện đáng kể sinh khả dụng và hoạt tính sinh học của EGCG đã được phát triển, bao gồm các loại hạt nano, nhũ tương nano và liposome nano khác nhau.
Một số nghiên cứu lâm sàng đã xác nhận tính an toàn của EGCG. Tuy nhiên, liều lượng cao (hơn 800 mg) có thể liên quan đến độc tính gan. Việc hàng ngày uống liều lượng bằng hoặc trên 800 mg EGCG có thể làm tăng đáng kể transaminase huyết thanh, trong khi lượng EGCG lên đến 704 mg/ngày được tiêu thụ dưới dạng đồ uống hoặc 338 mg EGCG/ngày được ăn vào dưới dạng bolus rắn cô đặc được coi là an toàn.
Resveratrol. Resveratrol tồn tại ở hai dạng đồng phân hình học – dạng trans và cis. Dạng cis phát sinh từ dạng trans bằng cách đồng phân hóa dưới ánh sáng UV và pH cao . Mặc dù nhìn chung, dạng trans của resveratrol được cho là có hoạt tính sinh học cao hơn, nhưng dạng cis cũng có thể có các đặc tính có lợi không giống với dạng đồng phân trans.
Mặc dù khoảng 70% resveratrol được hấp thụ và đạt nồng độ tối đa trong huyết tương là 2 µM sau khi dùng 25 mg, nhưng nó có sinh khả dụng thấp do quá trình chuyển hóa mạnh mẽ ở cả ruột và gan, dẫn đến sự kết hợp sulfat và axit glucuronic và hydro hóa liên kết đôi béo. Việc tăng liều lên tới 5 g dẫn đến sự gia tăng resveratrol không thay đổi lên tới 530 ng/mL. Để tăng sinh khả dụng của nó, một số công thức nano resveratrol đã được phát triển, ức chế hiệu quả sự phát triển của khối u.
Một số nghiên cứu lâm sàng đã cung cấp bằng chứng về sự an toàn của resveratrol. Các thử nghiệm lâm sàng ngẫu nhiên đã chỉ ra rằng việc hàng ngày bổ sung 500 mg resveratrol là an toàn và cải thiện chỉ số khối cơ thể và bài tiết insulin ở những bệnh nhân tiểu đường. Nói chung, resveratrol được dung nạp tốt ở liều lên đến 5 g/ngày; tuy nhiên, các tác dụng phụ nhẹ đến trung bình có thể xảy ra ở liều trên 1 g/kg. Một số nghiên cứu lâm sàng cho thấy bệnh nhân ung thư đại trực tràng, dạ dày và gan có thể hưởng lợi từ việc sử dụng resveratrol.
Curcumin. Là thành phần chính của củ nghệ, curcumin đã được con người sử dụng trong nhiều thiên niên kỷ và được gọi là “thần dược của cuộc sống”. Theo phân loại của Cục Quản lý Thực phẩm và Dược phẩm (FDA), nghệ được Công nhận Chung là An toàn (GRAS) và cho phép tiêu thụ curcumin với liều 3 mg/kg trọng lượng cơ thể. Do hoạt tính sinh học cao và độ an toàn phù hợp, curcumin hiện được bán tự do trên thị trường dưới dạng thực phẩm chức năng. Nó đã được nghiên cứu thành công trong hàng chục thử nghiệm lâm sàng để điều trị các bệnh khác nhau, bao gồm cả ung thư.
Nhược điểm của curcumin là sinh khả dụng thấp, do độ tan trong nước kém, khả năng hấp thụ đường tiêu hóa hạn chế, chuyển hóa nhanh, thanh thải toàn thân nhanh và khả năng thẩm thấu qua hàng rào máu não bị hạn chế.
Để cải thiện sinh khả dụng, một số tá dược (EGCG, piperine) và công thức nano curcumin (hạt nano, nhũ tương nano, nanocomposite, hydrogel) đã được phát triển, bao gồm cả những công thức đã được cấp bằng sáng chế và có sẵn trên thị trường. Ví dụ, curcumin ở dạng phức hợp galactomannoside (CurQfen®) có sinh khả dụng được cải thiện đáng kể, khả năng thấm qua hàng rào máu não và khả năng hấp thụ tế bào và chứng minh tính an toàn trong các thử nghiệm lâm sàng với liều lượng ~ 380 mg curcuminoid do các tình nguyện viên khỏe mạnh tiêu thụ trong 90 ngày.
Tổng hợp hoạt tính sinh học cao và độ an toàn phù hợp, có vẻ như curcumin có tiềm năng lớn như một chất bổ trợ trong liệu pháp chống ung thư.
Shikonin. Việc cho chuột Wistar ăn 200, 400 và 800 mg/kg shikonin trong vài tháng không gây ra bất kỳ độc tính nào. Ngoài ra, không có, hoặc nhỏ, tác dụng phụ đã được quan sát thấy trên chó Beagle ăn 100–2000 mg / kg shikonin trong 1–3 tuần. Hơn nữa, shikonin đã được sử dụng trong sáu thử nghiệm lâm sàng (tóm tắt trong), một nửa trong số đó nhằm mục đích điều trị ung thư và u xơ tử cung. Tuy nhiên, shikonin được báo cáo là một chất ức chế UGT (UDP-glucuronosyltransferases) có thể đảo ngược và có khả năng gây độc tính trong tương tác thuốc-thuốc hoặc thức ăn-thuốc. Nó cũng có thể ức chế các thành viên của họ cytochrome P450 . Nói chung, độc tính in vitro của shikonin cao hơn nhiều so với in vivo, điều này có thể là do sinh khả dụng thấp của nó.
Một số hệ thống mang thuốc nano đã được phát triển để cải thiện nó. Albreht và các đồng nghiệp đã chỉ ra rằng độ hòa tan của shikonin có thể tăng lên đến 181 lần khi thêm β-lactoglobulin. Mặt khác, việc đóng gói shikonin trong các hạt nano được phủ bằng saponin và sophorolipid đã cải thiện đáng kể độ hòa tan và sinh khả dụng của nó. Trong một nghiên cứu khác, shikonin được nạp vào micelles MPEG-PCL (methoxy poly (ethylene glycol)-b-poly (ε-caprolactone)) ức chế hiệu quả EMT trong các tế bào mô hình nội mô.
Hơn nữa, shikonin được bọc trong liposome đã thể hiện hoạt tính tăng lên in vivo (được xem xét trong). Do đó, liposome shikonin phủ axit hyaluronic đã được điều chế để nhắm mục tiêu hiệu quả các tế bào TNBC (ung thư vú âm tính ba) thông qua quá trình nội bào tử qua trung gian CD44. Trong một nghiên cứu khác, micelle ngụy trang màng chứa shikonin đã được phát triển để nhắm mục tiêu các khối u TNBC ở chuột.
Arctigenin. Arctigenin có sinh khả dụng kém và chuyển hóa lần đầu qua đường tiêu hóa rộng rãi. Việc uống arctigenin 70 mg/kg cho chuột dẫn đến nồng độ trong mô đạt đỉnh sau 30 phút và nhanh chóng bị đào thải trong vòng 4 giờ. Nồng độ arctigenin cao nhất được quan sát thấy ở lá lách, tiếp theo là gan và các cơ quan khác. Trong một thử nghiệm lâm sàng với những bệnh nhân ung thư tuyến tụy, liều uống chiết xuất từ quả cây ngưu bàng giàu arctigenin (GBS-01) với liều lượng 12 g arctigenin/người, nồng độ đỉnh của nó trong huyết tương là 66,56 ± 26,81 ng/mL, với AUC 487,97 ± 368,86 ng·h/mL.
Sự hấp thu, phân phối, chuyển hóa và đào thải arctigenin trong các mô hình in vitro và in vivo và các thử nghiệm lâm sàng được tóm tắt trong.
Sự an toàn của arctigenin vẫn chưa được nghiên cứu kỹ. Một công trình báo cáo rằng nó có thể gây độc đối với các tế bào không phải khối u ở vú. Tuy nhiên, một thử nghiệm lâm sàng giai đoạn I về chiết xuất từ quả cây ngưu bàng giàu arctigenin (GBS-01) đã được thực hiện trên 15 bệnh nhân ung thư tuyến tụy tiến triển kháng gemcitabine. Các tác giả báo cáo rằng liều 4 g/ngày GBS-01 dẫn đến đáp ứng lâm sàng thuận lợi và không có độc tính đáng kể. Hơn nữa, arctigenin cũng đã được nghiên cứu trong ba thử nghiệm lâm sàng khác, cho thấy hiệu quả của nó trong điều trị bệnh thận do tiểu đường.
Cucurbitacins. Cucurbitacin B là hợp chất được nghiên cứu nhiều nhất trong số các cucurbitacin, và do đó, thông tin tài liệu về sinh khả dụng và độ an toàn của hợp chất này là toàn diện nhất. Các thí nghiệm trên chuột Wistar đã chỉ ra rằng sinh khả dụng đường uống của cucurbitacin B là 10%, với nồng độ cao nhất trong huyết tương dao động từ 1 đến 100 ng/mL và đạt giá trị tối đa trong vòng khoảng 30 phút. Hơn nữa, cucurbitacin B thể hiện tỷ lệ mô/huyết tương cao, tích tụ gấp khoảng 10 lần trong một số cơ quan. Sự tích tụ tối đa của cucurbitacin được quan sát thấy ở phổi, lá lách và thận, tiếp theo là gan, dạ dày và ruột non, sau đó là não và tim. Việc chuột Wistar ăn 8 mg/kg cucurbitacin B dẫn đến nồng độ thuốc đỉnh (Cmax) là 34,16 ± 2,91 ng/L.
Như đã đề cập trước đó, cucurbitacin B ở dạng tinh khiết có liều gây chết trung bình ~5 mg/kg (đường uống) và 1 mg/kg (trong màng bụng) ở chuột nhắt, 0,5 mg/kg (tiêm tĩnh mạch) ở thỏ và 0,32 mg/kg (tiêm tĩnh mạch) ở mèo. Cucurbitacin B dưới dạng viên nén đã được sử dụng ở Trung Quốc như một chất bổ trợ để điều trị viêm gan mãn tính và ung thư gan từ những năm 1980. Một số nghiên cứu lâm sàng cho thấy cucurbitacin B làm tăng thời gian sống sót chung trong nhóm bệnh nhân ung thư gan, đồng thời làm giảm các triệu chứng lâm sàng liên quan đến viêm gan.
Theo như chúng tôi được biết, cho đến nay, chưa có nghiên cứu lâm sàng cụ thể nào về đặc điểm an toàn của cucurbitacin ở người được mô tả trong tài liệu. Cũng không có thông tin về độc tính cấp tính và kéo dài của nó. Do đó, cần phải có các nghiên cứu tiền lâm sàng và lâm sàng chuyên sâu để thiết lập hồ sơ an toàn và liều tối ưu của cucurbitacin B và các cucurbitacin khác ở động vật có vú, bao gồm cả con người.
Axit Betulinic. Do bản chất triterpene của nó, axit betulinic có sinh khả dụng kém. Tiêm trong màng bụng 500 mg/kg axit betulinic vào da chuột dẫn đến Cmax 300,9 µg/mL, thời gian bán hủy là 11,8 giờ, với sự tích tụ đáng kể ở buồng trứng, lá lách, tuyến vú, tử cung, bàng quang, hạch bạch huyết và gan.
Để cải thiện sinh khả dụng, các hệ thống phân phối BA khác nhau đã được tạo ra, bao gồm ống nano carbon, hạt nano từ tính và polyme, chất liên hợp, nhũ tương nano, liposome và cyclodextrin. Do đó, Saneja và các đồng nghiệp đã phát triển chất liên hợp BA-monomethoxy polyethylene glycol (mPEG), cải thiện đáng kể độ hòa tan của BA và hiệu quả chống khối u. BA-mPEG nội bào và gây ra quá trình apoptosis trong tế bào ung thư gan và vượt trội hơn đáng kể trong việc giảm khối u ở chuột ung thư cổ trướng Ehrlich so với BA không liên hợp. Ngoài ra, các dẫn xuất khác nhau của BA đã được tổng hợp và đánh giá. Ví dụ, các sửa đổi ở vị trí C-3, C-20 và C-28 có thể cải thiện độ hòa tan trong nước mà không ảnh hưởng đến hoạt tính dược lý của nó.
Theo một số nghiên cứu, BA không có tác dụng phụ đáng kể, chứng tỏ độc tính tế bào chọn lọc đối với tế bào ung thư. Nó đã được nghiên cứu trong một số thử nghiệm lâm sàng; tuy nhiên, các nghiên cứu lâm sàng của nó bị hạn chế trước hết là do độ hòa tan trong nước kém. Vì BA là một hợp chất chống ung thư rất hứa hẹn, chúng tôi đang chờ đợi các thử nghiệm lâm sàng liên quan đến liệu pháp điều trị dựa trên BA nano.
Ginsenosides. Ginsenosides được đặc trưng bởi quá trình chuyển hóa sinh học phức tạp trong và sau khi hấp thụ. Nhìn chung, chúng có độ hòa tan trong nước thấp, khả năng thấm qua màng kém và không ổn định về mặt trao đổi chất. Do đó, sinh khả dụng của chúng thấp và thường dưới 10% (dao động từ 0,3 đến 25%). Li và các đồng nghiệp đã chỉ ra rằng sau khi uống ginsenosides Rg1, Rb1 và Rd cho chuột cống, mức cao nhất của chúng được phát hiện ở gan, phổi, thận và lá lách.
Việc vi hóa ginsenoside Rh2 làm tăng sinh khả dụng của nó lên 32%. Hơn nữa, piperine có thể là một chất tăng cường hiệu quả sinh khả dụng của Rh2. Hơn nữa, rất nhiều công nghệ nano được áp dụng để tăng cường sinh khả dụng của ginsenosides và biến chúng thành liệu pháp chống ung thư.
Trong một thử nghiệm lâm sàng ngẫu nhiên, các tình nguyện viên đã nhận được chiết xuất hồng sâm hoặc hợp chất ginsenoside K (CK) với một phân tử glucose liên hợp (CK-30) – sản phẩm lên men của ginsenoside CK. Khi so sánh với chiết xuất hồng sâm, CK-30 cho thấy Cmax tăng 118,3 lần.
Các nghiên cứu khác nhau đã chỉ ra rằng ginsenosides được dung nạp tốt và có hồ sơ an toàn. Theo Song et al., việc hàng ngày uống 2 g hồng sâm Hàn Quốc trong 24 tuần là an toàn.
7. Kết luận và Triển vọng Tương lai
Trong bài đánh giá này, chúng ta đã thảo luận về đặc tính chống ung thư và cơ chế hoạt động của một số hợp chất tự nhiên được phân lập từ thực vật. Dựa trên việc sử dụng rộng rãi và tính đặc hiệu đa mục tiêu của chúng, các hợp chất này có thể có tiềm năng lớn để trở thành liệu pháp chống ung thư mới. Như đã đề cập trước đó, tất cả chúng đều nhắm mục tiêu vào nhiều con đường sinh hóa và tín hiệu và đáp ứng tiêu chí của liệu pháp đa mục tiêu. Hơn nữa, ngoài hoạt động chống khối u, tất cả các hợp chất này còn thể hiện các đặc tính dược lý có lợi khác nhau trong các mô khỏe mạnh, bao gồm chống viêm, bảo vệ tim mạch, thần kinh, gan, hạ đường huyết, v.v. Những đặc tính này có giá trị to lớn đối với bệnh nhân ung thư đang trải qua can thiệp hóa trị liệu vì họ chắc chắn sẽ phải đối mặt với tác dụng phụ của hóa trị và các hợp chất tự nhiên này có thể giúp phủ nhận những hậu quả không mong muốn.
Có thể suy ra từ các nghiên cứu trên rằng nhiều hợp chất trong số này có thể làm nhạy cảm tế bào ung thư với nhiều liệu pháp điều trị khác nhau. Một cách tiếp cận khả thi khác là kết hợp các hợp chất được mô tả để tăng cường tác động tiêu cực của chúng đối với nhiều con đường trao đổi chất và tín hiệu, thúc đẩy sự phát triển của các khối u ác tính.
Một trong những thách thức chính để đưa các hợp chất tự nhiên vào phòng khám là sinh khả dụng thấp của chúng. Tuy nhiên, có rất nhiều nỗ lực thành công được mô tả trong tài liệu để tăng sinh khả dụng của chúng bằng hệ thống phân phối nano hoặc chất tăng cường sinh học. Rõ ràng, những phát triển hơn nữa theo hướng này là hoàn toàn cần thiết để biến các hợp chất tự nhiên thành các liệu pháp có sẵn trên lâm sàng.
Tất cả các hợp chất được thảo luận đều thể hiện một loạt các đặc tính an toàn. Nhiều loại trong số chúng đã được nghiên cứu trong các thử nghiệm lâm sàng khác nhau hoặc/và được chấp thuận sử dụng làm thực phẩm bổ sung. Tuy nhiên, đối với phần lớn trong số chúng, không có đủ dữ liệu về độ an toàn và liều lượng của chúng để trở thành thuốc chống ung thư chính thức.
Bằng cách kết hợp phát triển các hệ thống phân phối nano phù hợp cho các hợp chất này với các nghiên cứu tiền lâm sàng và lâm sàng toàn diện, chúng tôi sẽ có thể giải phóng tiềm năng to lớn của các hợp chất tự nhiên để cải thiện nỗ lực của chúng tôi trong cuộc chiến chống ung thư.